

Nathalie Daher1; Thaissa Faloppa Duarte2; Gabriella Hiss Vetorasso3; Rafael Verardino Capalbo4; Thiago Martins Bachiega5
DOI: 10.17545/eoftalmo/2018.0002
ABSTRACT
Myeloid sarcoma is a rare, extramedullary solid tumor, which is usually associated with acute myeloid leukemia (AML), and may involve various parts of the body, including the eye socket. Its diagnosis is challenging and is mainly performed by anatomopathological and immunohistochemical analyses, because imaging findings are nonspecific. The objective of this study was to report the case of a patient in remission for AML, with a history of breast cancer and neurofibromatosis type 1, which evolved with rapid proptosis in the left eye and was diagnosed as myeloid sarcoma. Subsequent tests showed no recurrence of AML. This pathology has an indeterminate prognosis, and the main line of treatment includes chemotherapy, although radiotherapy and surgery may be used in selected cases.
Keywords: Sarcoma, Myeloid; Leukemia, Myeloid, Acute; Exophthalmos.
RESUMO
O Sarcoma Mieloide é um tumor extramedular sólido, raro, geralmente associado à leucemia Mieloide aguda (LMA), podendo envolver diversas partes do corpo, incluindo a órbita. Seu diagnostico é difícil e realizado principalmente pelo anatomopatológico e imunohistoquímica, pois os exames de imagem são inespecíficos. O objetivo deste trabalho é relatar o caso de uma paciente em remissão para LMA, com antecedentes de câncer de mama e neurofibromatose tipo 1, que evoluiu com rápida proptose em olho esquerdo, diagnosticada como sarcoma Mieloide. Os exames subsequentes não demonstraram recidiva da LMA. Essa patologia tem prognóstico indeterminado e sua principal terapêutica é a quimioterapia, embora radioterapia e cirurgia possam ser usadas em casos selecionados.
Palavras-chave: Sarcoma Mieloide; Leucemia Mieloide Aguda; Exoftalmia.
RESUMEN
El sarcoma mieloide es un tumor extramedular macizo, raro, en general asociado a casos de leucemia mieloide aguda (LMA), que puede presentarse en diferentes partes del cuerpo, incluyéndose la órbita. Su diagnóstico es difícil y se realiza sobre todo por los medios anatomopatológico y inmunohistoquímica, ya que los exámenes de imagen carecen de especificidad. El objetivo de este estudio es reportar el caso de una paciente en remisión para LMA, con caso anterior de cáncer de mama y neurofibromatosis tipo 1, que evolucionó con rápida proptosis en el ojo izquierdo, que se diagnosticó como sarcoma mieloide. Los exámenes subsecuentes no reportaron recidiva de la LMA. El pronóstico de esa patología es indeterminado y su principal terapia es la quimioterapia, aunque la radioterapia y la cirugía también sean opciones en casos apartados.
Palabras-clave: Sarcoma Mieloide; Leucemia Mieloide Aguda; Exofalmia.
INTRODUCTION
Myeloid sarcoma (MS), also known as granulocytic sarcoma or chloroma, is a rare, malignant solid tumor with extramedullary location and is formed by granulocyte precursors (myeloblasts)1.It is common in patients with acute myeloid leukemia (AML) but can also be associated with chronic leukemia; myeloproliferative neoplasms, such as polycythemia vera and myelofibrosis; and myelodysplastic syndrome2.
Any part of the body can be affected by MS; the most common locations include the soft tissues, bones, peritoneum, lymph nodes, skin, kidneys, and gastrointestinal system13. Less frequently affected sites include the eye socket, paranasal sinuses, and central nervous system3.
MS may precede, accompany, or indicate the recurrence of pretreated AML4. It is more common in children than in adults5. Its appearance in the remission phase of leukemia is a factor for poor prognosis, although cases of good evolution have been reported with early diagnosis and appropriate treatment21.
Thus, its suspected diagnosis is fundamental and is confirmed using anatomopathological studies and immunohistochemistry. The main treatment approach is usually chemotherapy19, 27.
Neurofibromatosis type 1 (Von Recklinghausen’s disease) is a generally congenital, hereditary, and autosomal dominant disease caused by a mutation in NF16, 29. The main clinical signs are café au lait spots, dermal and plexiform neurofibromas, axillary and inguinal freckling, and Lisch nodules12, 28. Some studies have demonstrated the association of neurofibromatosis type 1 with a higher incidence of some types of cancer, such as breast cancer, as reported in our patient22, 25.
Although MS is more frequent in other tissues, the importance of ocular involvement should not be overlooked; it has been described in this case report, which addresses the most relevant aspects of the disease that may be useful for its diagnosis and treatment.
CASE REPORT
A 47-year-old Caucasian female visited the Ophthalmology Department of the Redentora Eye Hospital due to hyperemia, pain of moderate intensity, and horizontal diplopia in the left eye, with an evolution period of 2 months (Figure 1). She had a history of neurofibromatosis type 1 diagnosed 32 years ago, left breast cancer treated with total mastectomy and chemotherapy 8 years ago, and AML type M3 (acute promyelocytic leukemia) in remission after chemotherapy and bone marrow transplant from a related donor 5 years ago. There was no significant family history.
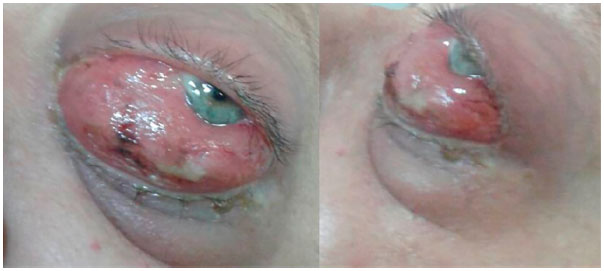
Ophthalmological examination showed visual acuity with correction 1.0 for distant vision and J1 for near vision in both the eyes. On ectoscopy, the right eye showed no changes, whereas the left eye showed hyperemia 3+/4+, conjunctival chemosis 4+/4+, palpebral edema 2+/4+, dystopia, and an axial proptosis of 7 mm, which did not increase with the Valsalva maneuver. Extrinsic ocular motility was decreased in the left eye. Biomicroscopy of the anterior segment showed the right eye without changes and left eye with hyperemia, conjunctival chemosis, and mild punctate exposure keratitis. There were no Lisch nodules (iris hamartomas), which are characteristic of neurofibromatosis type 1. Intraocular pressure was 13 mmHg in the right eye and 18 mmHg in the left eye, and fundoscopy findings and pupil reflexes were within the normal range. General physical examination showed three neurofibromas and six café au lait spots distributed on the trunk and limbs as well as axillary freckling due to neurofibromatosis type 1.
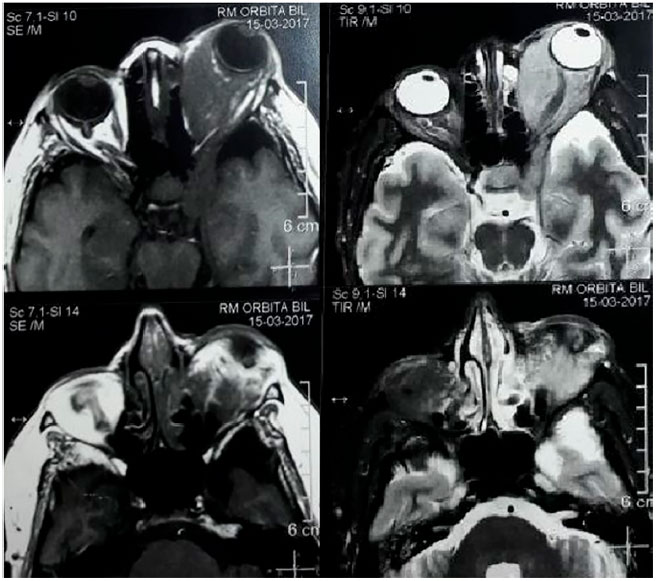
Hemogram, evaluation of T3, T4, and TSH (Thyoid Stimulator Hormone) levels, standard chest X-ray examination, and magnetic resonance imaging (MRI) of the brain and orbit were requested, and an oral corticosteroid (prednisone 40 mg/day) and topical ocular treatment were prescribed due to the nonspecific inflammatory process and lubricating drops due to exposure keratitis. On her follow-up visit, the hemogram, thyroid function test results, and chest X-ray findings were unchanged. MRI showed a normal right eye socket and left eye socket with a signal intensity mass similar to the soft parts which were isointense on T1- and hyperintense on T2-weighted images, with homogeneous intra- and extracoronal impregnation enhanced by the paramagnetic agent (gadolinium). The left cavernous sinus was also affected, with compression of the orbital plate and increased thickness of the extrinsic muscles (particularly the medial and inferior rectus muscles), optic nerve involved and with preserved diameter, and eyeball diverted anteriorly with normal sphericity and signal intensity (Figure 2). A lymphoproliferative lesion was suspected, and the patient was referred for biopsy of the lesion.
The anatomopathological report of the pathology service of the University of São Paulo (USP) in Ribeirão Preto (Serpat-HC) revealed left eye socket infiltration by a hematopoietic neoplasm composed of intermediate-sized neoplastic cells with a high nucleus/cytoplasm ratio, nuclei with irregular borders, moderate-intensity chromatin, immunohistochemically positive for CD117, CD34, myeloperoxidase and negative for CD20, CD3, CD5, CD23, and cyclin D1. MS was therefore diagnosed. A systemic investigation was conducted, including a new hemogram and bone marrow puncture, which showed no bone marrow involvement, thereby ruling out AML recurrence.
We opted for an exclusively chemotherapeutic treatment with three cycles of daunorubicin and cytarabin. The patient responded favorably to treatment, with a marked reduction in proptosis and all the eye conditions by the end of the last chemotherapy cycle (Figure 3).
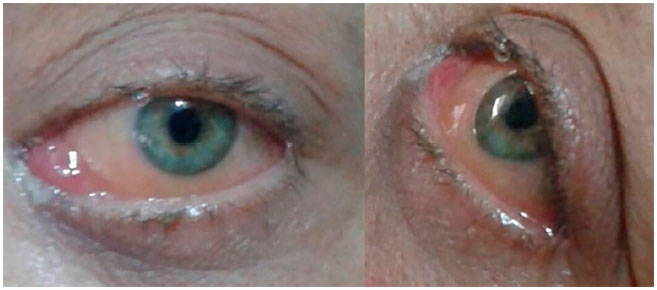
On follow-up ophthalmological examination at 10 months after the onset of symptoms, the patient had no further complaints and presented no relevant ocular changes. Her corrected visual acuity remained at 1.0 for distant vision and J1 for near vision. Her clinical picture evolved without further complications, and she is now in outpatient follow-up, asymptomatic, and without bone marrow involvement since the start of the eye complaints (11 months ago).
DISCUSSION
MS occurs concomitantly with AML at a rate of 2%–9%2, either as a single or multifocal tumor; it can occur either in association with other hematological diseases or in isolated form7. It is called chloroma due to its typical greenish coloration caused due to the exposure of the enzyme myeloperoxidase to ultraviolet light, although one-third of cases present other colors, such as white, gray, or red, depending on the predominant cells8. In most cases, it occurs following the diagnosis of AML but can also occur before or concomitantly with the AML diagnosis or indicate its recurrence in a patient previously treated for this hematological disease10.
Leukemias are malignancies derived from hematopoietic cells, which proliferate in the bone marrow24. Among the several risk factors for AML are chemotherapeutic agents, such as those used in breast cancer, previous radiotherapy, and inherited genetic syndromes, such as neurofibromatosis type 1. AML can lead to the development of extramedullary leukemic cell masses affecting various tissues. Ocular involvement is rare, and changes in the retina and choroid are predominant9. The eye socket is rarely involved, and its characteristic symptoms are proptosis, palpebral edema, and chemosis10.
First described in 1811 by Allen Burns, MS is also known as myeloblastoma, myelocytoma, chloroleukemia, and granulocytic sarcoma. It is composed of immature cells of the granulocytic series, originating from a myeloid precursor, usually myeloblasts11.
Orbital MS may appear at any age, but it is more common in children with a mean age of approximately 7 years. The disease is rare in adults10. It may affect any part of the body; the most common sites include the soft tissues, bones, peritoneum, lymph nodes, skin, kidneys, and gastrointestinal system13. Orbital involvement is rare and comprises 10% of orbital tumors along with neoplasms of lymphoid origin14.
The predilection for bone marrow or subperiosteum involvement in the axial skeleton may be related to increased hematopoiesis in these locations7, and cases affecting the eye socket can be explained by an origin in the adjacent bone, lacrimal gland, or extraocular muscles15.
The main signs of ocular involvement are pain, increased volume, involvement of adjacent tissues, inflammation of the eye socket, orbital tumors (palpebral and conjunctival), uveitis, and proptosis, the latter being the most common sign10.16. Systemic involvement can vary from 1 to 16 years after the appearance of the orbital lesion17.
The most commonly used immunohistochemical markers for the diagnosis of MS are positivity for MPO, CD68, CD43, lysozyme, CD45, CD117 (Ckit), CD99, CD33, CD34, and CD13 and negativity for CD3 and CD202.19. The most frequent cytogenetic alterations are similar to those found in AML, such as t(8.21) and inv(16). These changes are often associated with the orbital location of the tumor18.
The risk factors for the development of MS include cytogenetic abnormalities, such as t (8; 21), inv(16), AML subtypes FAB M4 and M5, increased white blood cells, presence of neural cell adhesion molecules (NCAM), T-cell markers (CD2, CD4, and CD7), poor nutrition, and low socioeconomic level19. MS is more common in AML subtypes M2, M4, and M519. Clinical suspicion of this tumor is difficult, and an initial AML presentation in a primarily extramedullary site with unaffected bone marrow and normal hematological findings is very rare20. When an orbital tumor is the initial manifestation, peripheral blood and bone marrow usually tend to be involved after 12 months without adequate treatment10.
Differential diagnoses for orbital MS include non-Hodgkin lymphoma, rhabdomyosarcoma, anaplastic carcinoma, lymphoblastic leukemia, Ewing sarcoma, eosinophilic granuloma, extramedullary hematopoiesis, melanoma, metastatic neuroblastoma, African Burkitt lymphoma, and idiopathic inflammatory pseudotumor5.
On computed tomography (CT), MS presents as iso-or hyperdense lesions with uniform contrast uptake23. On MRI, the lesions appear as hypo-or isointense on T1 and T2, with homogeneous contrast uptake and a tendency to remain isointense in sequences with a long repetition time (due to the presence of the myeloperoxidase enzyme)8.
The treatment of orbital MS without systemic involvement is not standardized, and its prognosis is still undetermined. The therapeutic choice for isolated MS is induction chemotherapy used in AML27. In MS associated with hematological disease, the treatment is focused on the underlying disease16. Some studies have demonstrated the superiority of systemic chemotherapy over local radiotherapy and surgery27.
Chemotherapy should be started even in cases of MS without bone marrow involvement19. Although MS is highly radiosensitive, radiotherapy does not increase the patient’s survival rate and is more appropriate in cases of poor response to chemotherapy and recurrence after hematopoietic bone marrow transplant. Surgery is reserved for rapid symptomatic relief, such as when there is compression of adjacent structures causing functional impairment27.
CONCLUSIONS
Although ophthalmologists have a secondary role in the diagnosis of primary hematological diseases, such as leukemia, their knowledge of these pathologies is of vital importance for the diagnosis of extramedullary diseases that might have more serious consequences to initiate early appropriate treatment.
The objective of this work was to present and discuss the clinical manifestations in a rare case of orbital MS without hematological or bone marrow involvement. Its prognosis has not been established and is highly variable. Early diagnosis and appropriate treatment, which usually includes chemotherapy, are very important.
Therefore, a multidisciplinary approach to MS is needed. Ophthalmologists must pay attention to its atypical clinical manifestations to deal with eye complications at an early stage and avoid systemic involvement.
REFERENCES
1. Cheah KL, Lim LC, Teong HH, Chua SH. A case of generalised cutaneous granulocytic sarcoma in an elderly patient with myelodysplastic syndrome. Singapore Med J. 2002;43(10):527-9.
2. Neiman RS, Barcos M, Berard C, et al. Granulocytic sarcoma: a clinicopathologic study of 61 biopsied cases. Cancer 1981;48:1426-1437.
3. Yilmaz AF, Saydam G, Sahin F, Baran Y. Granulocytic sarcoma:a systematic review. American Journal of Blood Research.2013;3(4):265–70.
4. Fonseca Junior NL, Paves L, Nakanami DM, Seixas MT, Manso PG. Sarcoma granulocítico em órbita: relato de caso. Arq Bras Oftalmol. 2005;68(4):557-60.
5. Pui MH, Fletcher BD, Langston JW. Granulocytic sarcoma in childhood leukemia: Imaging features. Radiology 1994;190:698-702.
6. Friedman, JM. Epidemiology of neurofibromatosis type 1. Am J Med Genet. 1999;89:1-6.
7. Deme S, Deodhare SS, Tucker WS, Bilbao JM. Granulocytic sarcoma of the spine in nonleukemic patients: report oh three cases. Neurosurgery 1997;40:1283-1287.
8. Parker K, Hardjasudarma M, McClellan RL, Fowler MR, Milner JW. MR features of an intracerebellar chloroma. Am J Neuroradiol 1996;17:1592-1594.
9. Kincaid MC, Green WR. Ocular and orbital involvement in leukemia. Surv Ophthalmol 1983;27(4):211.
10. Zimmerman LE, Font RL. Ophthalmologic manifestations of granulocytic sarcoma. Am J Ophthalmol 1975;80(6):975.
11. Pizzo PA, Poplack DG, editors. Principles and practice of pediatric oncology. 2nd ed. Philadelphia: J.B. Lippincott; 1993. p.483-500.
12. Tonsgard JH. Clinical manifestations and management of neurofibromatosis type 1. Semin Pediatr Neurol, 2006;13:2-7.
13. Liu PI, Ishimaru T, McGregor DH, Akada H, Steer A. Autopsy study of granulocytic sarcoma (chloroma) in patients with myelogenous leukemia, Hiroshima- Nagasaki 1949–1969. Cancer 1973;31:948–55.
14. Valvassori GE, Sabnis SS, Mafee RF, Brown MS, Putterman A. Imaging de distúrbios linfoproliferativos orbitários. Radiol Clin North Am 1999; 37: 135- 150.
15. Davis JL, Parke DW 2nd, Font RL. Granulocytic sarcoma of theorbit. A clinicopathologic study. Ophthamol.1985;92(12):1758–62.
16. Hetzler L T, Manera R, Lapentino S, Hotaling A. Primary granulocytic sarcoma presenting as an external auditory canal mass in a newborn with a draining ear. Int J Pediatr Otorhinolaryngol. 2009;4:1-5.
17. Mason TE, Demaree Jr RS, Margolis CI. Granulocytic sarcoma (chloroma), two years preceding myelogenous leukemia. Cancer. 1973;31(2):423-32.
18. Movassaghian, M., Brunner, A. M., Blonquist, T. M., Sadrzadeh, H., Bhatia, A., Perry, A. M., Attar, E. C., Amrein, P. C., Ballen, K. K., Neuberg, D. S., and Fathi, A. T. (2014) Presentation and outcomes among patients with isolated myeloid sarcoma: a Surveillance, Epidemiology, and End Results database analysis. Leuk. Lymphoma 56, 1–6.
19. Byrd JC, Edenfield WJ, Shields DJ, Dawson NA. Extramedullary myeloid cell tumors in acute nonlymphocytic leukemia: A clinical review. J Clin Oncol 1995;13:1800-16.
20. Scheinberg DA, Maslak P, Weiss M. Acute leukemias. In: Devita VT, Hellman S, Rosenberg SA (eds). Cancer: principles and practice of oncology. 5.ed. Philadelphia: Lippincott Raven; 1997, 2293-2316.
21. Infante AJ, MacRae MA, Forbes GS, Miller RH, Gilchrist GS. Intracranial granulocytic sarcoma complicating childhood acute myelomonocytic leukemia. Am J Pediatr Hematol Oncol 1981;3:173-176.
22. Sharif S, Moran A, Huson SM, Iddenden R, Shenton A, Howard E, Evans DGR. Women with neurofibromatosis 1 are at a moderately increased risk of developing breast cancer and should be considered for early screening. J Med Genet 2007: 44, 481-4.
23. Barnett MJ, Zussman WV. Granulocytic sarcoma of the brain: a case report and review of the literature. Radiology 1986;160:223-225.
24. Scheinberg DA, Golde DW. The leukemias, Harrison.s. Principles of Internal Medicine, 13th ed, McGraw-Hill, 1994, cap 310.
25. Walker L, Thompson D, Easton D, Ponder B, Ponder M, Frayling I, Baralle D. A prospective study of neurofibromatosis typ1 cancer incidence in the UK. Br J Cancer 2006: 95, 232-8.
26. Ruggieri M, Huson SM. The neurofibromatosis: a overview. Ital J Neurol Sci. 1999;20:89-108.
27. Yamauchi, K., and Yasuda, M. (2002) Comparison in treatments of nonleukemic granulocytic sarcoma: Report of two cases and a review of 72 cases in the literature. Cancer 94, 1739–1746.
28. Ellis BD. Optic pathway gliomas. In: Tasman W, Jaeger EA. Duane's Ophthalmology 2006 Edition (CD-ROM). Philadelphia: Lippincott Willians & Wilkins; 2005. cap 42.
29. Ferner RE. Neurofibromatosis 1 and neurofibromatosis2: a twenty first century perspective. Lancet Neurol, 2007;6:340-351.
30. North K. Clinical aspects of neurofibromatosis 1. Eur J Paediatr Neurol. 1998;2:223-31.
Funding: No specific financial support was available for this study.
CEP Approval: Not applicable.
Disclosure of potential conflicts of interest: None of the authors have any potential conflict of interest to disclose.
Received on:
December 17, 2017.
Accepted on:
March 15, 2018.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados





 Ler em português
Ler em português
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket