

Nathalie Daher1; Thaissa Faloppa Duarte2; Gabriella Hiss Vetorasso3; Rafael Verardino Capalbo4; Thiago Martins Bachiega5
DOI: 10.17545/eoftalmo/2018.0002
RESUMO
O Sarcoma Mieloide é um tumor extramedular sólido, raro, geralmente associado à leucemia Mieloide aguda (LMA), podendo envolver diversas partes do corpo, incluindo a órbita. Seu diagnostico é difícil e realizado principalmente pelo anatomopatológico e imunohistoquímica, pois os exames de imagem são inespecíficos. O objetivo deste trabalho é relatar o caso de uma paciente em remissão para LMA, com antecedentes de câncer de mama e neurofibromatose tipo 1, que evoluiu com rápida proptose em olho esquerdo, diagnosticada como sarcoma Mieloide. Os exames subsequentes não demonstraram recidiva da LMA. Essa patologia tem prognóstico indeterminado e sua principal terapêutica é a quimioterapia, embora radioterapia e cirurgia possam ser usadas em casos selecionados.
Palavras-chave: Sarcoma Mieloide; Leucemia Mieloide Aguda; Exoftalmia.
ABSTRACT
Myeloid sarcoma is a rare, extramedullary solid tumor, which is usually associated with acute myeloid leukemia (AML), and may involve various parts of the body, including the eye socket. Its diagnosis is challenging and is mainly performed by anatomopathological and immunohistochemical analyses, because imaging findings are nonspecific. The objective of this study was to report the case of a patient in remission for AML, with a history of breast cancer and neurofibromatosis type 1, which evolved with rapid proptosis in the left eye and was diagnosed as myeloid sarcoma. Subsequent tests showed no recurrence of AML. This pathology has an indeterminate prognosis, and the main line of treatment includes chemotherapy, although radiotherapy and surgery may be used in selected cases.
Keywords: Sarcoma, Myeloid; Leukemia, Myeloid, Acute; Exophthalmos.
RESUMEN
El sarcoma mieloide es un tumor extramedular macizo, raro, en general asociado a casos de leucemia mieloide aguda (LMA), que puede presentarse en diferentes partes del cuerpo, incluyéndose la órbita. Su diagnóstico es difícil y se realiza sobre todo por los medios anatomopatológico y inmunohistoquímica, ya que los exámenes de imagen carecen de especificidad. El objetivo de este estudio es reportar el caso de una paciente en remisión para LMA, con caso anterior de cáncer de mama y neurofibromatosis tipo 1, que evolucionó con rápida proptosis en el ojo izquierdo, que se diagnosticó como sarcoma mieloide. Los exámenes subsecuentes no reportaron recidiva de la LMA. El pronóstico de esa patología es indeterminado y su principal terapia es la quimioterapia, aunque la radioterapia y la cirugía también sean opciones en casos apartados.
Palabras-clave: Sarcoma Mieloide; Leucemia Mieloide Aguda; Exofalmia.
INTRODUÇÃO
O sarcoma mieloide (SM), também conhecido como sarcoma granulocítico ou cloroma, é um tumor sólido maligno raro, de localização extramedular, formado por precursores de granulócitos (mieloblastos)1.É frequente em pacientes com leucemia mieloide aguda (LMA), mas também pode estar associado a leucemias crônicas, neoplasias mieloproliferativas (como policitemia vera e mielofibrose) e síndrome mielodisplásica2.
Qualquer parte do corpo pode ser acometida pelo SM, sendo os locais mais comuns tecidos moles, ossos, peritônio, linfonodos, pele, rins e sistema gastrointestinal13. Menos frequentes são a orbita, seios paranasais e sistema nervoso central3.
Pode anteceder, acompanhar ou indicar recorrência de uma LMA já tratada4, e é mais frequente em crianças do que em adultos5. Seu surgimento na fase de remissão das leucemias é fator de mau prognóstico, embora tenham sito relatados casos de boa evolução com diagnóstico precoce e tratamento adequado21.
Dessa forma, sua suspeita diagnóstica é fundamental, sendo confirmada com estudos anatomopatológicos e a imunohistoquímica, e seu tratamento costuma ter como abordagem principal a quimioterapia19,27.
Já a neurofibromatose tipo 1 (doença de Von Reckinghausen) é uma doença congênita, hereditária e autossômica dominante, causada por uma mutação no gene NF16,29. Os principais sinais clínicos são as mancha café-com-leite, os neurofibromas dérmicos e plexiformes, sardas axilares e inguinais e nódulos de Lisch12,28. Alguns estudos demonstraram a associação da neurofibromatose tipo 1 com maior incidência de alguns tipos de cânceres, como câncer de mama, relatado pela nossa paciente22,25.
Embora o SM seja mais frequente em outros tecidos, o acometimento ocular não deve ter sua importância diminuída, sendo descrito neste relato de caso, que abordou os aspectos mais pertinentes à doença que podem ser úteis para o seu diagnóstico e tratamento.
RELATO DE CASO
Paciente do sexo feminino, 47 anos, branca, compareceu ao serviço de oftalmologia do HO Redentora Hospital de Olhos referindo hiperemia, dor de intensidade moderada e diplopia horizontal em olho esquerdo, com tempo de evolução de dois meses (figura 1). Tinha como antecedentes pessoais neurofibromatose tipo 1 diagnosticada há 32 anos, câncer de mama esquerda tratado com mastectomia total e quimioterapia há 8 anos e leucemia mieloide aguda (LMA) tipo M3 (leucemia promielocítica aguda) em remissão após quimioterapia e transplante de medula óssea de doador relacionado há 5 anos. Sem antecedentes familiares significativos.
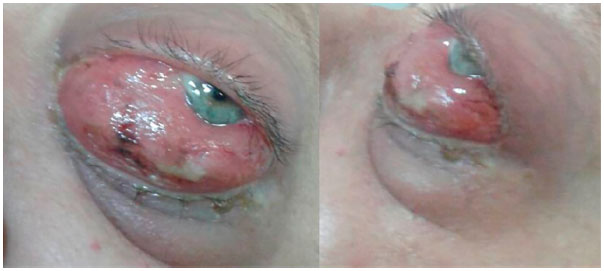
Ao exame oftalmológico, apresentava acuidade visual com correção 1,0 para longe e J1 para perto em ambos os olhos. Na ectoscopia, olho direito sem alterações e olho esquerda com hiperemia 3+/4+, quemose conjuntival 4+/4+, edema palpebral 2+/4+, distopia e proptose axial de 7 mm que não aumentava com manobra de Valsalva. A motilidade ocular extrínseca estava diminuída em olho esquerdo. Biomicroscopia do segmento anterior evidenciou olho direito sem alterações e olho esquerdo com hiperemia, quemose conjuntival e ceratite punctata leve de exposição. Não havia presença de nódulos de Lisch (hamartomas irianos), característicos da neurofibromatose tipo 1. A pressão intraocular era 13 mmHg em olho direito e 18mmHg em olho esquerdo, e a fundoscopia e os reflexos pupilares encontravam-se dentro da normalidade. O exame físico geral relevou três neurofibromas e seis manchas café com leite distribuídos em tronco e membros, além de sardas axilares, devido à neurofibromatose tipo 1.
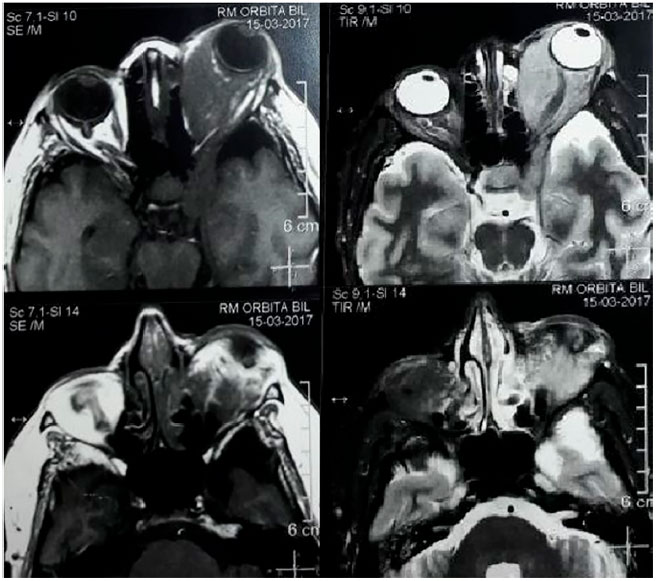
Foi solicitado hemograma, T3, T4L, TSH (hormônio estimulante da tireóide), radiografia simples de tórax, ressonância magnética (RM) cerebral e de órbita, e iniciado corticoterapia via oral (prendnisona 40mg/dia) e tópica ocular, devido ao processo inflamatório inespecífico, além de colírios lubrificantes para a ceratite de exposição. No retorno, o hemograma e as provas de funções tireiodianas estavam sem alterações, assim como a radiografia torácica. A RM revelou órbita direita dentro da normalidade e órbita esquerda com massa de intensidade de sinal semelhante a partes moles, isointensa em T1 e hiperintensas em T2 com acentuada impregnação homogênea pelo agente paragnético (gadolíneo), intra e extracoronal, acometendo também o seio cavernoso esquerdo, com compressão da lâmina papirácea e aumento de espessura dos músculos extrínsecos (principalmente retomedial e retoinferior), nervo óptico envolvido e com diâmetro preservado, e globo ocular desviado anteriormente com esfericidade e intensidade de sinal normais (figura 2). Suspeitou-se de lesão linfoproliferativa e a paciente foi encaminhada para biópsia da lesão.
O laudo do anatomopatológico do serviço de patologia da USP (Universidade de São Paulo) de Ribeirão Preto (Serpat-HC) revelou órbita esquerda com infiltração por neoplasia hematopoiética constituída por células neoplásicas de tamanho intermediário, com alta relação núcleo/citoplasma, núcleos de contornos irregulares e cromatina de moderada intensidade, com positividade imunohistoquimica para CD117, CD34, mieloperoxidase, e negatividade para CD20, CD3, CD5, CD23 e ciclina D1; desta forma, diagnosticou-se sarcoma mieloide. Foi realizada investigação sistêmica, incluindo novo hemograma e punção de medula óssea, que não demonstraram acometimento medular, descartando-se recidiva da LMA.
Optou-se pelo tratamento quimioterápico exclusivo, com três ciclos de daunorubicina e citarabina. A paciente respondeu favoravelmente ao tratamento, com acentuada redução da proptose e de todo quadro ocular já no final do último ciclo quimioterápico (figura 3).
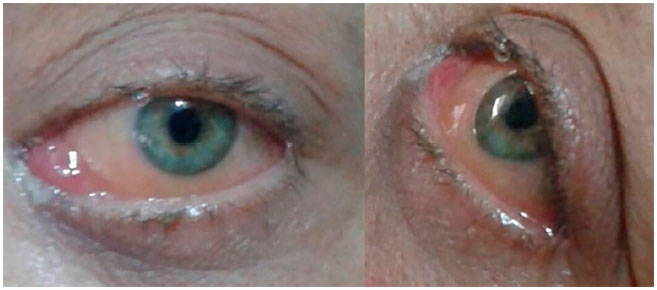
Em novo exame oftalmológico, após 10 meses do início do quadro, a paciente não tinha novas queixas nem apresentava alterações oculares relevantes, e sua acuidade visual com correção permanecia 1,0 para longe e J1 para perto. Seu quadro evoluiu sem intercorrências e a mesma encontra-se em seguimento ambulatorial, assintomática no momento e sem acometimento medular desde o início do quadro ocular, há 11 meses.
DISCUSSÃO
O sarcoma mieloide ocorre concomitante à leucemia mielóide aguda (LMA) na frequência de 2% a 9%2, como um tumor único ou multifocal, como também pode ocorrer associado a outras doenças hematológicas ou de forma isolada7. É chamado de cloroma por sua típica coloração esverdeada, devido à presença da enzima mieloperoxidase ao se expor na luz ultravioleta, embora um terço dos casos apresente outras cores, como esbranquiçada, acinzentada e avermelhada, de acordo com as células predominantes8. Na maioria das vezes ocorre após o diagnóstico de LMA, mas também pode anteceder, ser concomitante ou indicar sua recidiva em um paciente previamente tratado desta doença hematológica10.
Leucemias são neoplasias derivadas de células hematopoiéticas, que proliferam-se a partir da medula óssea24. Dentre os diversos fatores de risco para LMA encontram-se quimioterápicos (como os utilizados em câncer de mama), radioterapia prévia, e síndromes genéticas hereditárias como neurofibromatose tipo 1. A LMA pode causar o desenvolvimento de massas de células leucêmicas extramedulares com acometimento de diversos tecidos. É raro o acometimento ocular, onde predomina alterações na retina e coroide9. A órbita é pouca envolvida, e seu quadro característico é com proptose, edema palpebral e quemose10.
Descrito primeiramente em 1811 por Allen Burns, o SM também é denominado de mieloblastoma, mielocitoma, cloroleucemia e sarcoma granulocítico, e é composto de células imaturas da série granulocítica, a partir de precursor mieloide, usualmente mieloblastos11.
O sarcoma mieloide da órbita pode apresentar-se em qualquer faixa etária, mas é mais frequente em crianças com idade média de aproximadamente sete anos, sendo o acometimento de pacientes adultos mais raro10. Pode afetar qualquer local do corpo, sendo os mais comuns tecidos moles, ossos, peritônio, linfonodos, pele, rins e sistema gastrointestinal13. É raro o envolvimento orbitário, e juntamente com as neoplasias de origem linfóide, constituem 10% dos tumores orbitais14.
A predileção pelo envolvimento ósseo ou subperióstico no esqueleto axial pode estar relacionada a maior hematopoiese nestes locais7, e os casos orbitais podem ser explicados pela sua origem a partir de osso adjacente, glândula lacrimal ou músculos extraoculares15.
O acometimento ocular tem como principais sinais dor, aumento de volume e envolvimento de tecidos adjacentes, inflamação orbitaria, tumorações oculares (como palpebral e conjuntival), uveíte e proptose, sendo este ultimo o sinal mais comum10,16. O acometimento sistêmico pode variar de 1 a 16 anos após o surgimento da lesão orbitaria17.
Os marcadores imunohistoquimicos mais utilizados no diagnóstico do SM são positividade para MPO, CD68, CD43, lisosima, CD45, CD117 (Ckit), CD99, CD33, CD34 e CD13, e negatividade para CD3 e CD202,19. As alterações citogenéticas mais frequentes são semelhantes às encontradas na LMA, como t(8,21) e inv 16, alterações mais associadas à localização orbitária do tumor18.
Fatores de risco para o desenvolvimento do SM são anormalidades citogenéticas como t (8; 21), inv (16), subtipos FAB M4 e M5 da LMA, aumento de glóbulos brancos, presença de moléculas de adesão celular neural (NCAM), marcadores de células T (CD 2, CD 4, CD 7), nutrição deficiente e baixo nível socioeconômico19. O SM é mais comum nos subtipos M2, M4 e M5 da LMA19.
A suspeita clínica deste tumor é difícil, e a apresentação inicial de LMA em um sítio primariamente extramedular com medula óssea não acometida e exames hematológicos normais é muito rara20. Quando o tumor orbital é a manifestação inicial, o sangue periférico e o medula óssea costuma costumam ser envolvidos após 12 meses sem tratamento adequado10.
Diagnósticos diferenciais para o SM orbitário são linfoma não hodgkin (LNH), rabdomiossarcoma, carcinoma anaplasico, leucemia linfoblastica, sarcoma de Ewing, granuloma eosinofílico, hematopoiese extramedular, melanoma, neuroblastoma metastático, linfoma de Burkitt Africano e pseudotumor inflamatório idiopático5.
Na tomografia computadorizada (TC), o SM apresenta-se como lesões iso ou hiperdensas, com captação uniforme de contraste23, e na ressonância magnética (RM), as lesões são hipo ou isointensas em T1 e T2, com captação homogênea de contraste e tendência a permanecer isointensos em sequências com longo tempo de repetição (pela presença da enzima mieloperoxidase)8.
O tratamento de SM orbitário sem acometimento sistêmico não é padronizado e seu prognóstico ainda é indeterminado. A escolha terapêutica para SM isolado é a quimioterapia de indução utilizada na LMA27, e no SM associado à doença hematológica, o tratamento é voltado para a doença de base16. Alguns estudos demonstraram superioridade da quimioterapia sistêmica, em relação à radioterapia local e cirurgia27.
A quimioterapia deve ser iniciada ainda nos casos de SM sem comprometimento da medula óssea19. Embora o SM seja altamente radiossensível, a radioterapia não aumenta a sobrevida do paciente é mais apropriada em casos de pouca resposta á quimioterapia e recorrência após transplante hematopoiético de medula óssea, e a cirurgia é reservada para alivio sintomático rápido, como em compressão de estruturas adjacentes causando déficits funcionais27.
Alguns trabalhos relacionaram a neurofibromatose tipo 1 a maior incidência de câncer. O primeiro trabalho foi em 2006, por Walker et al, revelando o risco de câncer 2,7 vezes maior em pacientes com neurofibromatose do que na população em geral, com risco cumulativo de câncer aos 50 anos de 20%, demonstrando que o risco de câncer de mama foi significativamente maior nas mulheres com menos de 50 anos portadoras de neurofibromatose do que na população em geral25. Em 2007, Sharif et al realizaram um estudo mostrando um risco de câncer de mama 5 vezes maior para mulheres com neurofibromatose do que em mulheres sem neurofibromatose22.
Até o momento não há tratamento eficaz para prevenir ou retardar as lesões da neurofibromatose tipo 1, e sua terapêutica é voltada para manejar as complicações tratáveis, além do aconselhamento genético12.
CONCLUSÕES
Embora o oftalmologista tenha um papel secundário primário no diagnostico de doenças hematológicas, como as leucemias, seu conhecimento sobre essas patologias é de vital importância para fazer o diagnostico de doenças extramedulares que possam ter repercussões mais graves, a fim de se iniciar um tratamento adequado precocemente.
O objetivo deste trabalho foi apresentar e discutir as manifestações clínicas num caso raro de SM orbitário sem envolvimento hematológico ou de medula óssea. Seu prognóstico é indeterminado e muito variável, sendo de vital importância seu diagnóstico precoce e tratamento adequado, geralmente com quimioterapia.
Portanto, o SM deve compreender uma abordagem multidisciplinar. Os oftalmologistas devem ficar atentos às suas manifestações clinicas atípicas, a fim de manejar precocemente as complicações oculares e evitar o comprometimento sistêmico do paciente.
REFERÊNCIAS
1. Cheah KL, Lim LC, Teong HH, Chua SH. A case of generalised cutaneous granulocytic sarcoma in an elderly patient with myelodysplastic syndrome. Singapore Med J. 2002;43(10):527-9.
2. Neiman RS, Barcos M, Berard C, et al. Granulocytic sarcoma: a clinicopathologic study of 61 biopsied cases. Cancer 1981;48:1426-1437.
3. Yilmaz AF, Saydam G, Sahin F, Baran Y. Granulocytic sarcoma:a systematic review. American Journal of Blood Research.2013;3(4):265–70.
4. Fonseca Junior NL, Paves L, Nakanami DM, Seixas MT, Manso PG. Sarcoma granulocítico em órbita: relato de caso. Arq Bras Oftalmol. 2005;68(4):557-60.
5. Pui MH, Fletcher BD, Langston JW. Granulocytic sarcoma in childhood leukemia: Imaging features. Radiology 1994;190:698-702.
6. Friedman, JM. Epidemiology of neurofibromatosis type 1. Am J Med Genet. 1999;89:1-6.
7. Deme S, Deodhare SS, Tucker WS, Bilbao JM. Granulocytic sarcoma of the spine in nonleukemic patients: report oh three cases. Neurosurgery 1997;40:1283-1287.
8. Parker K, Hardjasudarma M, McClellan RL, Fowler MR, Milner JW. MR features of an intracerebellar chloroma. Am J Neuroradiol 1996;17:1592-1594.
9. Kincaid MC, Green WR. Ocular and orbital involvement in leukemia. Surv Ophthalmol 1983;27(4):211.
10. Zimmerman LE, Font RL. Ophthalmologic manifestations of granulocytic sarcoma. Am J Ophthalmol 1975;80(6):975.
11. Pizzo PA, Poplack DG, editors. Principles and practice of pediatric oncology. 2nd ed. Philadelphia: J.B. Lippincott; 1993. p.483-500.
12. Tonsgard JH. Clinical manifestations and management of neurofibromatosis type 1. Semin Pediatr Neurol, 2006;13:2-7.
13. Liu PI, Ishimaru T, McGregor DH, Akada H, Steer A. Autopsy study of granulocytic sarcoma (chloroma) in patients with myelogenous leukemia, Hiroshima- Nagasaki 1949–1969. Cancer 1973;31:948–55.
14. Valvassori GE, Sabnis SS, Mafee RF, Brown MS, Putterman A. Imaging de distúrbios linfoproliferativos orbitários. Radiol Clin North Am 1999; 37: 135- 150.
15. Davis JL, Parke DW 2nd, Font RL. Granulocytic sarcoma of theorbit. A clinicopathologic study. Ophthamol.1985;92(12):1758–62.
16. Hetzler L T, Manera R, Lapentino S, Hotaling A. Primary granulocytic sarcoma presenting as an external auditory canal mass in a newborn with a draining ear. Int J Pediatr Otorhinolaryngol. 2009;4:1-5.
17. Mason TE, Demaree Jr RS, Margolis CI. Granulocytic sarcoma (chloroma), two years preceding myelogenous leukemia. Cancer. 1973;31(2):423-32.
18. Movassaghian, M., Brunner, A. M., Blonquist, T. M., Sadrzadeh, H., Bhatia, A., Perry, A. M., Attar, E. C., Amrein, P. C., Ballen, K. K., Neuberg, D. S., and Fathi, A. T. (2014) Presentation and outcomes among patients with isolated myeloid sarcoma: a Surveillance, Epidemiology, and End Results database analysis. Leuk. Lymphoma 56, 1–6.
19. Byrd JC, Edenfield WJ, Shields DJ, Dawson NA. Extramedullary myeloid cell tumors in acute nonlymphocytic leukemia: A clinical review. J Clin Oncol 1995;13:1800-16.
20. Scheinberg DA, Maslak P, Weiss M. Acute leukemias. In: Devita VT, Hellman S, Rosenberg SA (eds). Cancer: principles and practice of oncology. 5.ed. Philadelphia: Lippincott Raven; 1997, 2293-2316.
21. Infante AJ, MacRae MA, Forbes GS, Miller RH, Gilchrist GS. Intracranial granulocytic sarcoma complicating childhood acute myelomonocytic leukemia. Am J Pediatr Hematol Oncol 1981;3:173-176.
22. Sharif S, Moran A, Huson SM, Iddenden R, Shenton A, Howard E, Evans DGR. Women with neurofibromatosis 1 are at a moderately increased risk of developing breast cancer and should be considered for early screening. J Med Genet 2007: 44, 481-4.
23. Barnett MJ, Zussman WV. Granulocytic sarcoma of the brain: a case report and review of the literature. Radiology 1986;160:223-225.
24. Scheinberg DA, Golde DW. The leukemias, Harrison.s. Principles of Internal Medicine, 13th ed, McGraw-Hill, 1994, cap 310.
25. Walker L, Thompson D, Easton D, Ponder B, Ponder M, Frayling I, Baralle D. A prospective study of neurofibromatosis typ1 cancer incidence in the UK. Br J Cancer 2006: 95, 232-8.
26. Ruggieri M, Huson SM. The neurofibromatosis: a overview. Ital J Neurol Sci. 1999;20:89-108.
27. Yamauchi, K., and Yasuda, M. (2002) Comparison in treatments of nonleukemic granulocytic sarcoma: Report of two cases and a review of 72 cases in the literature. Cancer 94, 1739–1746.
28. Ellis BD. Optic pathway gliomas. In: Tasman W, Jaeger EA. Duane's Ophthalmology 2006 Edition (CD-ROM). Philadelphia: Lippincott Willians & Wilkins; 2005. cap 42.
29. Ferner RE. Neurofibromatosis 1 and neurofibromatosis2: a twenty first century perspective. Lancet Neurol, 2007;6:340-351.
30. North K. Clinical aspects of neurofibromatosis 1. Eur J Paediatr Neurol. 1998;2:223-31.
Fonte de financiamento: declaram não haver.
Parecer CEP: não aplicável.
Conflito de interesses: declaram não haver.
Recebido em:
17 de Dezembro de 2017.
Aceito em:
15 de Março de 2018.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados





 Ler em inglês
Ler em inglês
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket