

Simone Cristine Hermes Hoff1; Aline Christina Fioravanti Lui2; Eliza Antonieta Rosana Negrão Grangeiro3
DOI: 10.17545/e-oftalmo.cbo/2016.63
RESUMO
A epiteliopatia pigmentar placoide multifocal posterior aguda (EPPMPA) é uma doença incluída na síndrome dos pontos brancos; é rara e autolimitada, cursa com baixa de acuidade visual (BAV) súbita e múltiplas lesões placoides, bilateral e assimétrica. Descreve-se aqui um caso de uma paciente de 50 anos, com BAV súbita (AV AO: CD a 1 m), com achados fundoscópicos e angiofluoresceinográficos muito semelhantes à Síndrome de Vogt-Koyanagi-Harada, com múltiplas áreas de descolamento de retina, bilaterais, assimétricas, envolvendo a área macular e a periferia. A diferenciação diagnóstica foi feita baseada na ausência de pan-uveíte e/ou achados extraoculares, e boa AV final (20/30 AO), mesmo na ausência de tratamento.
Palavras-chave: Coroidite; Síndrome Uveomeningoencefálica; Angiofluoresceinografia; Descolamento Retiniano
ABSTRACT
Acute posterior multifocal pigment epitheliopathy (apmppe) is classified as white dot syndrome, It is rare and self-limited and involves low visual acuity and sudden multiple placoid lesions, which are bilateral and asymmetrical, The case of a 50 year-old patient with sudden visual loss was reported (visual acuity in both eyes: counting the number of fingers at 1 meter), with alterations in the fundus and fluorescein angiography results similar to that observed in vogt-koyanagi-harada syndrome, with multiple areas of bilateral, asymmetric retinal detachment involving the macular area and periphery, Differential diagnosis was made based on the absence of panuveitis and/or extraocular findings, and good final visual acuity (20/30 in both eyes) was observed, even without treatment.
Keywords: Choroiditis; Uveomeningoencephalitic Syndrome; Fluorescein angiography; Retinal detachment
INTRODUÇÃO
A síndrome dos pontos brancos compreende um heterogêneo grupo de síndromes caracterizadas por múltiplas lesões em polo posterior com inflamação da coroide, epitélio pigmentar da retina e retina. 1
Dentre as doenças incluídas nessa síndrome existe a epiteliopatia pigmentar placoide multifocal posterior aguda (EPPMPA), que é uma doença rara e autolimitada, acometendo tipicamente adultos jovens (20-50 anos). 2,3 Essa epiteliopatia foi descrita originalmente por Gass, em 1968, como uma patologia que primariamente afeta o EPR, de etiologia e patogenia pouco conhecida, de início abrupto, com múltiplas lesões placoides bilaterais e assimétricas, conferindo uma acuidade visual (BAV) súbita.1 O tratamento tradicional constitui-se de corticoterapia oral por 3 a 6 semanas, evoluindo com bom prognóstico visual final.1
RELATO DE CASO
Paciente do sexo feminino, 50 anos, branca, natural e procedente de São Paulo, deu entrada no pronto-socorro com queixa de BAV súbita em ambos os olhos (AO) há 2 dias. Negava hipertensão arterial sistêmica, diabetes, cefaleia, zumbido e antecedentes de patologia oftalmológica.
Ao exame apresentava AV c/c CD a 1m AO, biomicroscopia e PIO normais.
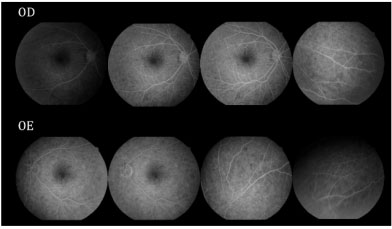
Retinografia da admissão evidenciou múltiplas áreas de descolamento de retina, bilateral, assimétrica, acometendo a área macular e a periferia da retina (Figuras 1 e 2).
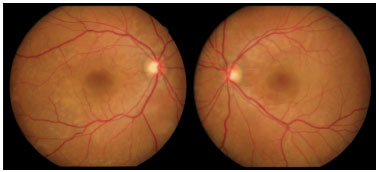
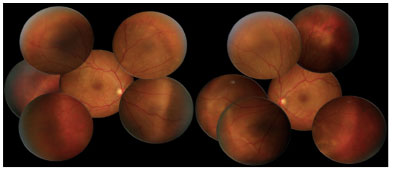
Na Angiofluoresceinografia (AFG) foram observadas múltiplas áreas de hiperfluorescência tardia bilateral, assimétrica, envolvendo a área macular e periferia da retina, correspondendo a DR seroso e descolamento de EPR. Fase tardia com múltiplas áreas de hipofluorescência (Figura 3).
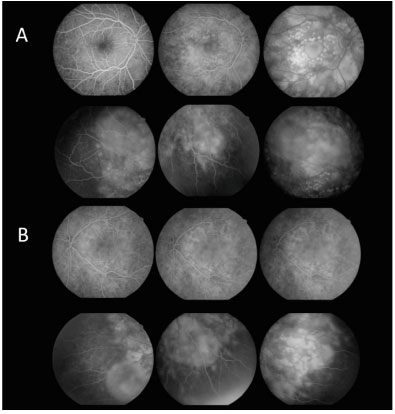
Foram solicitados hemograma, VHS, FR, PPD, sorologias para toxoplasmose, VDRL e FTAbs, Rx tórax, TC tórax e crânio, e todos apresentaram resultado dentro da normalidade. Foi prescrito prednisona VO 1 mg/Kg/dia.
Após 12 dias a paciente retorna com AV c/c 20/30 AO, sem fazer uso da medicação prescrita. Foi realizada retinografia com 12 e 30 dias de evolução, que evidenciou melhora importante do quadro, seguida de resolução espontânea (Figura 4). AFG com 30 dias de evolução apresentou múltiplas áreas puntiformes de hipofluorescência por defeito em janela, correspondendo a cicatriz perimacular com resolução dos múltiplos descolamentos de retina na área macular e periferia da retina (Figura 5).
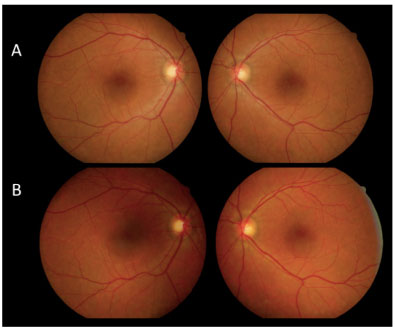
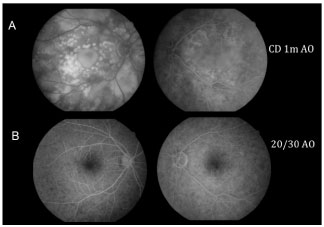
A figura 6 compara a AFG da área macular de ambos os olhos no início do quadro, com AV AO CD a 1m, e após 30 dias sem tratamento, com boa AV final (20/30 AO), na ausência de tratamento.
DISCUSSÃO
A EPPMPA é uma doença rara que cursa com múltiplas lesões placoides bilaterais e assimétricas, conferindo uma BAV súbita. 3 Apresenta, na AFG, hipo ou hiperfluorescência precoce, com hiperfluorescência que aumenta de intensidade e tamanho por extravasamento do contraste na fase tardia.4,5
A síndrome de Vogt-Koyanagi-Harada (SVKH) tem provável etiologia autoimune, que se manifesta com múltiplos descolamentos retinianos, bilaterais e assimétricos, resultando em perda visual grave se não tratada adequadamente.6 A síndrome completa apresenta alterações neurológicas, auditivas e cutâneas, além de pan-uveíte, sem história prévia de doença ocular, cirurgia ou trauma.7 Na AFG observam-se áreas de hiperfluorescência na fase precoce, com extravasamento na fase tardia decorrentes da presença de acúmulo de líquido sub-retiniano.6
Quando a EPPMPA cursa com áreas de descolamentos serosos de retina, a diferenciação diagnóstica fica dificultada pela semelhança dos achados angiofluoresceinográficos entre SVKH e EPPMA. A paciente relatada teve o diagnóstico confirmado de EPPMPA por não ter apresentado pan-uveíte e outras alterações extraoculares, e por ter evoluído de forma favorável com boa acuidade visual final (20/30 AO), e melhora das alterações fundoscópicas e angiofluoresceinográficas, apesar de não ter realizado nenhum tratamento.
REFERÊNCIAS
1. Gross NE, Yannuzzi LA, Freund KB, Spaide RF, Amato GP, Sigal R. Multiple evanescent white dot syndrome. Arch Ophthalmol. 2006;124(4):493-500. http://dx.doi.Org/10.1001/archopht.124.4.493
2. Durrani A, Rahimy E, Dunn JP. Bilateral acute-onset vision loss in a previously healthy man. JAMA Ophthalmol. 2015;133:957-8. http://dx.doi.org/10.1001/jamaophthalmol.2015.1139
3. Mrejen S, Gallego-Pinazo R, Wald KJ, Freund KB. Acute posterior multifocal placoid pigment epitheliopathy as a choroidopathy: what we learned from adaptive optics imaging. JAMA Ophthalmol. 2013;131:1363-4. http://dx.doi.org/10.1001/jamaophthalmol.2013.4196
4. Fine HF, Spaide RF, Ryan Jr EH, Matsumoto Y, Yannuzzi LA. Acute zonal occult outer retinopathy in patients with multiple evanescent white dot syndrome. Arch Ophthalmol. 2009;127:66-70. http://dx.doi.org/10.1001/archophthalmol.2008.530
5. Yeh S, Forooghian F, Wong WT, Faia LJ, Cukras C, Lew JC, et al. Fundus autoflourescence imaging of the white dot syndromes. Arch Ophthalmol. 2010;128:46-56. http://dx.doi.org/10.1001/archophthalmol.2009.368
6. Mota LAA, Santos AB. Síndrome de Vogt-Koyanagi-Harada e o seu acometimento multissistêmico. Rev Assoc Med Bras. 2010; 56(5):590-5. http://dx.doi.org/10.1590/S0104-42302010000500023
7. Lucena DR, Paula JS, Silva GCM, Rodrigues MLV. Síndrome de Vogt-Koyanagi-Harada incompleta associada a HLA DRB1*01 em criança de quatro nos de idade: relato de caso. Arq Bras Oftalmol. 2007;70(2):340-2. http://dx.doi.org/10.1590/S0004-27492007000200027



Fonte de financiamento: declaram não haver.
Conflito de interesses: declaram não haver.
Recebido em:
24 de Outubro de 2016.
Aceito em:
24 de Novembro de 2016.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados
 Ler em inglês
Ler em inglês
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket