

João Marçal Medeiros de Sousa1; Clayton Leite de Moura1; Hanniman Denizard Cosme Barbosa1; Thales Matheus Lopes de Sousa1; Lisley Medeiros Garcia1; Gabriela de Araújo Miranda1; Aganeide Castilho Palitot2
DOI: 10.17545/eOftalmo/2022.0002
RESUMO
OBJETIVOS: Neuromielite óptica é uma doença imunomediada cujo diagnóstico baseia-se na detecção do anticorpo anti-aquaporina 4. A neuromielite óptica é reconhecidamente associada a uma gama de doenças autoimunes.
MÉTODOS: Trata-se de uma revisão integrativa da literatura dos últimos 20 anos que busca investigar a relação entre a neuromielite óptica e o escleroderma sistêmico e a esclerodermia localizada. Incluiu-se artigos em português, inglês ou espanhol das bases de dados MEDLINE, Lilacs, SciELO e Scopus encontrados através das palavras-chave “Neuromielite óptica”, “Escleroderma Sistêmico” e “Esclerodermia Localizada”, seus sinônimos e correspondentes em inglês e espanhol.
RESULTADOS: A busca gerou 23 artigos. Três relatos de caso, um em inglês e dois e português, compuseram o corpus final deste trabalho.
DISCUSSÃO: A relação entre Neuromielite óptica e doenças autoimunes é estabelecida. Em relação ao escleroderma sistêmico e a esclerodermia localizada há pouca associação dentro da literatura, maioria oriunda de casos isolados, dando a esta associação o status de raridade. A baixa testagem, status sub-clínicos/iniciais de algumas apresentações da neuromielite óptica e terapias imunossupressoras podem explicar esta baixa associação.
CONCLUSÃO: Estudos de busca ativa e mensuração do anti-aquoporina 4 em pacientes com afecções auto-imunes se fazem necessários. Trata-se de um grande campo de pesquisa ainda a ser desbravado.
Palavras-chave: Neuromielite óptica; Escleroderma sistêmico; Esclerodermia localizada; Aquaporina 4; Anticorpo.
ABSTRACT
OBJECTIVES: To assess whether systemic and localized sclerosis have a significant correlation with neuromyelitis optica and its spectrum of diseases, aiming to open new study perspectives for these autoimmune diseases.
METHODS: This is an integrative review of the literature of the last 20 years, which aims to investigate the relationship between neuromyelitis optica, systemic scleroderma, and localized scleroderma. Articles in Portuguese, English, and Spanish from the MEDLINE, LILACS, SciELO, and Scopus databases were searched using the keywords “neuromyelitis optica,” “systemic scleroderma,” and “localized scleroderma,” along with their synonyms and correspondents in Portuguese and Spanish.
RESULTS: The search generated 23 articles. Three case reports, one in English and two in Portuguese, comprised the final corpus of this work.
DISCUSSION: The relationship between neuromyelitis optica and autoimmune diseases is well established, but its association with systemic and localized scleroderma is rarely reported in the literature. Furthermore, the reported associations primarily arise from isolated cases, thereby making this association relatively rare. Uncommon testing, the subclinical or initial status of some neuromyelitis optica presentations, and immunosuppressive therapies may explain this rare association.
CONCLUSION: Active search and measurement studies of anti-aquaporin 4 in patients with autoimmune disorders are necessary. This is a large field of research that requires further investigation.
Keywords: Neuromyelitis optica; Systemic scleroderma; Localized scleroderma; aquaporin 4; Antibody.
INTRODUÇÃO
A neuromielite óptica (NMO) é uma doença inflamatória autoimune desmielinizante de graves repercussões clínicas1. Suas lesões são predominantemente no nervo óptico e na medula espinal, poupando normalmente o cérebro, diferente da esclerose múltipla2, a qual já foi considerada uma variante localizada da doença1. Até 2004 seu diagnóstico era baseado em critérios menores, maiores e paraclínicos3, entretanto a descoberta de um anticorpo contra o canal de aquaporina 4 (NMO-IgG) mudou a abordagem da doença: a imunoglobulina passou a ser considerada seu principal agente etiológico, com elevada especificidade e sensibilidade4, possibilitando diagnóstico precoce, além de melhor tratamento e prognóstico a estes pacientes1.
A aquaporina 4 é o grande canal de água do sistema nervoso central (SNC). Está localizada nos astrócitos, sendo responsável pelo balanço hídrico cerebral e controle dos níveis séricos de potássio e glutamato2,5,6. A ligação do anticorpo anti-aquaporina 4 leva a um down regulation nestes canais, causando dano astrocitário e consequentemente neuronal, gerando processos inflamatórios via complemento e imunidade celular, desmielinização e edema no SNC6,7. Ademais, discute-se que o NMO-IgG aumenta a permeabilidade da barreira hematoencefálica permitindo que anticorpos, granulócitos e antígenos específicos de células T acessem esse ambiente outrora impermeável8.
Vale ressaltar que a descoberta do NMO-IgG abriu um novo campo de estudos, pois o mesmo anticorpo foi encontrado em outras doenças auto-imunes, criando-se o conceito de Desordens do Espectro da NMO (DENMO)1,9. As DENMO incluem a miastenia gravis, hipotireoidismo, anemia perniciosa, esclerodermia, colite ulcerativa, colangite esclerosante primária, púrpura trombocitopênica idiopática, síndrome de Sjögren, sarcoidose, síndrome do anticorpo antifosfolípide e lúpus eritematoso sistêmico10,11. O possível mecanismo de associação entre essas doenças autoimunes ainda é algo sob intensa investigação científica12.
A esclerodermia é uma dessas entidades nosológicas sob investigação nesse panorama. Trata-se de uma afecção imunomediada do tecido conjuntivo com grande espectro de apresentação clínica, sendo caracterizada pela esclerose resultante da deposição de colágeno13. Pode afetar somente pele, fáscia, músculo e ossos, sendo caracterizada como localizada (EL), ou se expandir e acometer órgãos internos, sendo denominada esclerose sistêmica (ES)14.
Várias são as fisiopatologias propostas para explicar a doença. Um das mais aceitas é a de que células endoteliais progenitoras disfuncionais circulantes exacerbariam a deterioração vascular gerando esclerose vascular e, portanto, sistêmica15.
Estudos recentes têm apontado que desregulação na expressão de aquoporinas em fibroblastos dérmicos e no endotélio da microvasculatura também podem ser responsáveis pela fibrose observada nestas afecções. O canal de aquaporina 1, por exemplo, participa da angiogênese e do remodelamento da matriz extracelular, encontrando-se expresso em grande intensidade em pacientes com ES16.
Outra situação via fisiopatológica é a dos canais de aquoporina 3. Estes, por sua vez, tem como papel participar do processo de cicatrização e desidratação da pele, diretamente relacionados à fisiopatologia da ES, de tal modo que nesses pacientes foi observado a subexpressão desses canais17.
Além disso, no passado, o envolvimento neurológico em pacientes com ES ou EL foi considerado algo raro e incidental, entretanto novos trabalhos têm apontado que cerca de 40% desses pacientes possuem esta associação, variando desde leves cefaleias até miopatias severas. Teoriza-se que a atividade inflamatória da ES poderia contribuir para o dano ao sistema nervoso central observado nas DENMO18.
Reconhecendo as doenças como detentoras de fisiopatologias complexas sabidamente associadas a mecanismos imunológicos e inflamatórios17, receptores da mesma família, de estrutura e funções semelhantes, bem como ainda desconhecidos, este trabalho visa avaliar se, da mesma forma como ocorre em outras doenças autoimunes e imunomediadas, a esclerose sistêmica e localizada, seriam doenças de correlação importante com a NMO/DENMO de modo a abrir novas perspectivas de estudo entre ambas as doenças autoimunes.
MÉTODOS
Trata-se de uma revisão de literatura do tipo integrativa. Para a seleção dos artigos foram utilizadas as bases de dados MEDLINE (Sistema Online de Busca e Análise de Literatura Médica), SciELO (Scientific Electronic Library Online), Lilacs (Literatura Latino-Americana e do Caribe em Ciências da Saúde) e Scopus. Inclui-se todos os artigos publicados em português, inglês ou espanhol nos últimos vinte anos. As palavras-chave utilizadas foram “Neuromielite Óptica”, “Escleroderma Sistêmico” e “Esclerodermia Localizada” (e seus respectivos sinônimos e termos em inglês e espanhol). Foram incluídos estudos clínicos, relatos de caso, estudo de coorte e caso-controle. Revisões integrativas, narrativas, cartas ao editor e artigos indisponíveis na íntegra foram excluídos.
RESULTADOS
A busca foi realizada e gerou em um total de 23 artigos (7 artigos no buscador BVS e 16 na base de dados Scopus). Após a exclusão de duplicatas, deu-se a fase de leitura dos artigos obtidos, eliminando-se aqueles que fugiam ao tema ou não perfaziam os critérios de inclusão estabelecidos (Figura 1).
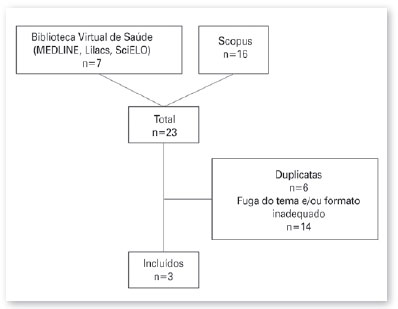
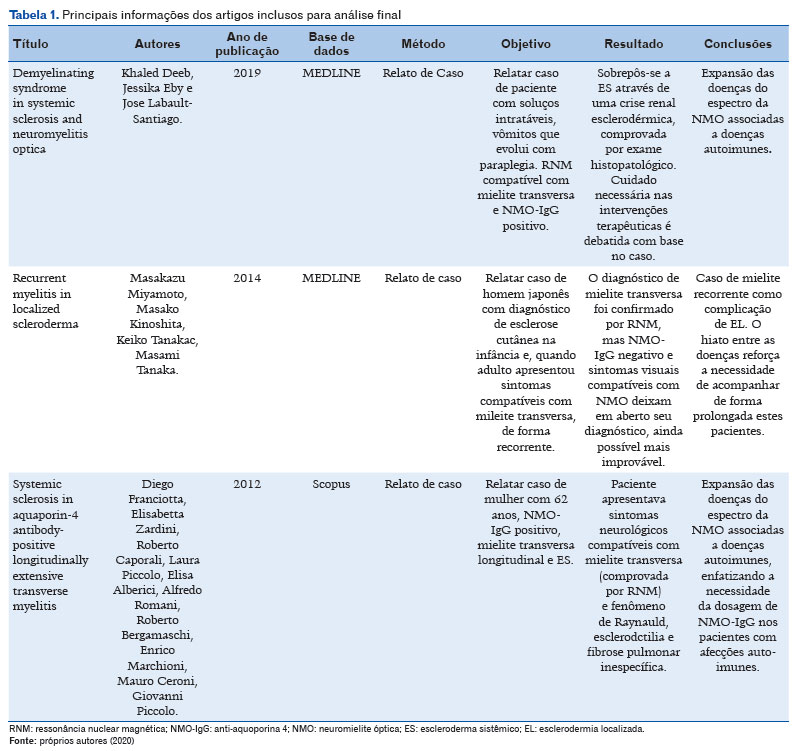
Por fim, foram selecionados 3 artigos, cujos principais dados estarão resumidos na tabela 1 a seguir.
DISCUSSÃO
Cerca de 20-30% dos portadores de NMO albergam outras afecções auto-imunes20, das quais se destacam lúpus eritematoso sistêmico, síndrome de Sjogren, artrite reumatoide, miastenia gravis e colite ulcerativa como as de maior correlação1, mas sua associação ao escleroderma sistêmico e a morfeia, ou esclerodermia localizada, é algo infrequente. Esse trabalho concentrou esforços em elucidar essa lacuna.
Três relatos de caso constituem parte do material final de estudo desta revisão. O primeiro deles trata-se de uma mulher de 44 anos cuja queixa principal eram a instalação aguda de soluços, vômitos e disfagia intratáveis. Notou-se que a paciente apresentava espessamento cutâneo nas junções metacarpo falangeanas bilateralmente e fenômeno de Raynaud. A posteriori a paciente desenvolveu turvação visual, fraqueza muscular, hipertonia, espasmos tônicos paroxísticos, sinal de Babinski e parestesia em membros inferiores. Ressonância nuclear magnética (RNM) demonstrou focos hiperintensos na substância branca periventricular e na medula espinhal entre C2 e C6. Anti-corpo anti-núcleo, anti-RNA polimerase III, topoisomerases I e NMO-IgG testaram positivos, concluindo-se, portanto, o diagnóstico de NO. Iniciou-se metilprednisolona intravenoso, trazendo significativa melhora clínica. No quinto dia de internação a paciente apresentou crise hipertensiva e injúria renal aguda, que exigiu hemodiálise de urgência. Biópsia foi performada conferindo o diagnóstico de crise renal esclerodérmica induzida pelo corticoide. Paciente foi tratada posteriormente com outros medicamentos, respondendo bem a rituximab18.
Outro caso trata-se de uma paciente 39 anos, sexo feminino, com antecedente de DENMO há 12 anos quando apresentou tetraplegia, RNM com mielite transversa cervical e AQP4 positivo. À ocasião evoluiu positivamente após pulsoterapia de metilprednisolona e uso de azatioprina. Foi então encaminhada para avaliação reumatológica devido espessamento cutâneo e fenômeno de Raynaud há um ano. Detectou-se ao exame físico telangiectasias em face, leucomelanodermia em tórax, microulcerações digitais e espessamento cutâneo difuso, com escore cutâneo de Rodnan modificado (mRSS) de 2217.
Apresentou Fator anti-núcleo nucleolar >1/1280, anti-Scl70 negativo, capilaroscopia periungueal padrão SD. Afastou-se envolvimento visceral, sendo então firmado o diagnosticado de ES difusa pelos critérios ACR/EULAR (2013). Foi introduzida, então, ciclofosfamida endovenosa mensal e, após oito meses de tratamento, houve melhora cutânea significativa17.
Por fim, o último trabalho analisado trata-se de um paciente HCCP, 54 anos, tabagista, hipertenso e submetido a prostatectomia por neoplasia há 2 anos. Há cerca de oito meses apresentou esclerodermia em placas, em membros inferiores, superiores e abdome de rápida evolução. Biópsia cutânea compatível com morfeia. Seis meses após avaliação inicial apresenta quadro de paraparesia de membros inferiores, constipação e retenção urinária, com nível sensitivo em T3, força grau 0 em membros inferiores, com Babinski em extensão bilateral. Sem qualquer queixa ocular. RNM demonstrou lesão desmielinizante em medula espinhal e Anti-NMO foi positivo, firmando o diagnóstico de DENMO. Fez-se pulsoterapia com metilprednisolona 1g por 5 dias, ciclofosfamida 1g, imunoglobulina 2g/kg e plasmaférese. Após 14 dias de internação observou-se melhora leve dos sintomas19.
A quantidade de artigos viáveis para seção final desta revisão, bem como a maioria se tratar de relatos de caso, é um indicador do status de raridade de pesquisas acerca da associação entre NMO, escleroderma sistêmico e a esclerodermia localizada. Outros estudos ratificam esse ineditismo, a exemplo do trabalho brasileiro realizado com 22 pacientes portadores de NMO: apenas um paciente apresentou o fenômeno de Raynauld, nenhuma descrição de escleroderma sistêmico ou esclerodermia localizada. A positividade de anti-corpos NMO-IgG dentro da amotra foi o mais significativo, como era de se esperar. Entretanto o anti-nucleosomo, anti-núcleo, anti-tireoperoxidase e anti-tireoglobulina, ficaram reservados a um número reduzido de pacientes23. Em uma extensa revisão de literatura que recuperou artigos desde 1976 a 2017 que tratavam de doenças auto-imunes associadas à NMO: apenas três casos de ES/EL foram localizados24.
Concluindo a presente revisão integrativa entendendo-se que o NMO-IgG está presente em diferentes e variadas doenças autoimunes e imunomediadas, entretanto a descrição da associação entre NMO, escleroderma sistêmico e a esclerodermia localizada é algo pouco observado na literatura médica disponível pesquisada, a nível de raridade. Este fato pode ser explicado pela baixa testagem de NMO-IgG realizada em pacientes com outras doenças auto-imunes, bem como a checagem de outras doenças auto-imunes em pacientes portadores de NMO23. Ademais, como observados nos casos relatados, por vezes a manifestação destas patologias pode ocorrer com anos de diferença19,24, e, mesmo havendo suspeita, esta pode não apresentar todos os critérios necessários para seu diagnóstico, seja pelo uso de outras medicações imunossupressoras20, comum nestes pacientes, seja pelo estágio bastante inicial da NMO em que se encontram, sem soropositividade ou manifestações clínicas exuberantes24.
Frente aos resultados apontados nos artigos incluídos nesta revisão integrativa entende-se como importante intensificar esforços para o desenvolvimento de mais trabalhos com delineamentos que sejam capazes de capturar mais dados além do alvo de análise neste presente escrito. Também seria prudente e oportuna a pesquisa da associação da NMO com outras patologias autoimunes e imunomediadas, haja vista o vasto campo de estudo e pesquisa disponível.
REFERÊNCIAS
1. Freitas E, Guimarães J. Neuromyelitis optica spectrum disorders associated with other autoimmune diseases. Rheumatol Int. 2015;35(2):243-53.
2. Sellner J, Boggild M, Clanet M, Hintzen RQ, Illes Z, Montalban X, et al. EFNS guidelines on diagnosis and management of neuromyelitis optica. Eur J Neurol. 2010;17(8):1019-32.
3. Fazio R, Radaelli M, Furlan R. Neuromyelitis optica: concepts in evolution. J Neuroimmunol. 2011;231(1-2):100-4.
4. Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet. 2004;364(9451):2106-12.
5. Jacob A, McKeon A, Nakashima I, Sato DK, Elsone L, Fujihara K, et al. Current concept of neuromyelitis optica (NMO) and NMO spectrum disorders. J Neurol Neurosurg Psychiatry. 2013; 84(8):922-30.
6. González C, González-Buitrago JM, Izquierdo G. Aquaporins, anti-aquaporin-4 autoantibodies and neuromyelitis optica. Clin Chim Acta. 2013;415:350-60.
7. Misu T, Höftberger R, Fujihara K, Wimmer I, Takai Y, Nishiyama S, et al. Presence of six different lesion types suggests diverse mechanisms of tissue injury in neuromyelitis optica. Acta Neuropathol. 2013;125(6):815-27.
8. Ingegnoli F, Gualtierotti R, Pierro L, Del Turco C, Miserocchi E, Schioppo T, et al. Choroidal impairment and macular thinning in patients with systemic sclerosis: the acute study. Microvasc Res. 2015;97:31-6.
9. Pittock SJ, Lennon VA, De Seze J, Vermersch P, Homburger HA, Wingerchuk DM, et al. Neuromyelitis optica and non-organ-specific autoimmunity. Arch Neurol. 2008;65(1):78-83.
10. Jarius S, Jacobi C, De Seze J, Zephir H, Paul F, Franciotta D, et al. Frequency and syndrome specificity of antibodies to aquaporin-4 in neurological patients with rheumatic disorders. Mult Scler J. 2011;17(9):1067-73.
11. Kolfenbach JR, Horner BJ, Ferucci ED, West SG. Neuromyelitis optica spectrum disorder in patients with connective tissue disease and myelitis. Arthritis Care Res (Hoboken). 2011;63(8): 1203-8.
12. Wingerchuk DM, Weinshenker BG. The emerging relationship between neuromyelitis optica and systemic rheumatologic autoimmune disease. Mult Scler J. 2012;18(1):5-10.
13. Zulian F, Cuffaro G, Sperotto F. Scleroderma in children: an update. Curr Opin Rheumatol. 2013;25(5):643-50.
14. Arango C, Malagón C, Gómez M del P, Mosquera C, Yépez R, González T, et al. Esclerodermia localizada juvenil:¿ es una enfermedad benigna? Rev Colomb Reumatol. 2017;24(3):145-52.
15. Trojanowska M. Cellular and molecular aspects of vascular dysfunction in systemic sclerosis. Nat Rev Rheumatol. 2010; 6(8):453.
16. Yamashita T, Asano Y, Saigusa R, Taniguchi T, Nakamura K, Miura S, et al. Increased expression of aquaporin-1 in dermal fibroblasts and dermal microvascular endothelial cells possibly contributes to skin fibrosis and edema in patients with systemic sclerosis. J Dermatol Sci. 2019;93(1):24-32.
17. Farhadi E, Mahmoudi M, Rahmani F, Yousefi B, Sarafnejad A, Kavosi H, et al. Attenuation of aquaporin-3 and epidermal growth factor receptor expression and activation in systemic sclerosis dermal fibroblasts. J Cell Physiol. 2019;234(8):12876-83.
18. Amaral TN, Peres FA, Lapa AT, Marques-Neto JF, Appenzeller S. Neurologic involvement in scleroderma: a systematic review. In: Seminars in arthritis and rheumatism. Elsevier; 2013. p. 335-47.
19. Teixeira JMA, Oliveira JL, Cordeiro RA, Luppino-Assad AP, Sampaio-Barros PD. Doença do espectro da neuromielite óptica e esclerose sistêmica: uma rara associação. Rev Bras Reumatol. 2017;57:S89-90.
20. Iyer A, Elsone L, Appleton R, Jacob A. A review of the current literature and a guide to the early diagnosis of autoimmune disorders associated with neuromyelitis optica. Autoimmunity. 2014;47(3):154-61.
21. Deeb K, Eby J, Labault-Santiago J. Demyelinating syndrome in systemic sclerosis and neuromyelitis optica. BMC Neurol. 2019; 19(1):1-6.
22. Maciel ML, Sa AT, Zimmermann AF, Neves FS, Fialho S, Pereira IA. Doença do espectro da neuromielite óptica com anti-aquaporina-4 associado a morfeia (esclerodermia localizada). Rev Bras Reumatol. 2017;57:S264-5.
23. Pereira WLDCJ, Reiche EMV, Kallaur AP, Oliveira SR, Simão ANC, Lozovoy MAB, et al. Frequency of autoimmune disorders and autoantibodies in patients with neuromyelitis optica. Acta Neuropsychiatr. 2017;29(3):170-8.
24. Shahmohammadi S, Doosti R, Shahmohammadi A, Mohammadianinejad SE, Sahraian MA, Azimi AR, et al. Autoimmune diseases associated with neuromyelitis optica spectrum disorders: a literature review. Mult Scler Relat Disord. 2019;27:350-63.
INFORMAÇÃO DOS AUTORES







Financiamento: Declaram não haver
Conflitos de Interesse: Declaram não haver
Recebido em:
24 de Junho de 2021.
Aceito em:
24 de Maio de 2022.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados
 Ler em inglês
Ler em inglês
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket