

Carlos Augusto Moreira Neto1; Letícia Costa Almeida Mariuzzo1; Vera Lúcia Rodrigues Falcão1; José Fábio de Oliveira Miranda1; Paula Maria Haggi Fregadolli1; Lunna Pires Moreira2
DOI: 10.17545/eOftalmo/2020.0018
RESUMO
A Síndrome de Bardet-Biedl é uma rara doença autossômica recessiva caracterizada por: polidactilia, obesidade, retardo mental, hipogenitalismo e distrofia retiniana. Alguns dos achados acima podem estar ausentes, mas a distrofia retiniana é achado consistente.WBR, 21 anos, masculino, procura atendimento para acompanhamento de retinite pigmentosa diagnosticado desde a infância. Possui irmão com o mesmo diagnóstico. Acuidade visual de conta dedos em ambos os olhos. Ao exame de fundo de olho apresenta disco óptico pálido, estreitamento de vasos, atrofia de epitélio pigmentar da retina e alterações pigmentares com aspecto de espículas ósseas na periferia. Ao exame físico, apresenta cicatrizes de correção da hexadactilia de mãos, obesidade, déficit auditivo e cognitivo. Além disso, possui diagnóstico prévio de hipogonadismo. Campimetria computadorizada mostrou-se não confiável em ambos os olhos. Observou-se então, que além de retinite pigmentosa o paciente se enquadrava nos critérios de Síndrome de Bardet-Biedl. A síndrome de Bardet-Biedl é uma rara doença de etiologia ainda incerta, com apenas poucos casos descritos na literatura. Embora atualmente não haja tratamento, o diagnóstico precoce é importante para o acompanhamento multidisciplinar, melhorando o desempenho em suas atividades diárias.
Palavras-chave: Distrofias retinianas; Retinose pigmentar; Síndrome de Bardet-Biedl.
ABSTRACT
Bardet-Biedl syndrome (BBS) is a rare autosomal recessive disease that is typically characterized by the following symptoms: polydactyly, obesity, mental retardation, hypogenitalism, and retinal dystrophy. Despite the absence of some of the abovementioned symptoms, retinal dystrophy is consistently observed in BBS patients. WBR, a 21-year-old man, seeked care for follow-up of pigmentary retinosis that was diagnosed since childhood. He has a brother with the same diagnosis. Count finger visual acuity was used to evaluate both eyes. Fundus examination revealed a pale optic disc, narrowing of vessels, atrophy of retinal pigment epithelium, and pigmentary changes with the appearance of bone spicules on the periphery. Physical examination revealed the correction scars related to the treatment of hexadactilia of both hands, along with other symptoms such as obesity, auditory, and cognitive deficits. In addition, he had previously been diagnosed with hypogonadism. Computerized campimetry proved to be unreliable in both eyes. It was observed that in addition to pigmentary retinosis, the patient met the diagnostic criteria of BBS. BBS is a rare disease with a currently unknown etiology, and there have only been a few cases of this condition have been reported in the literature. Although there is currently no treatment, early diagnosis is important for multidisciplinary follow-up to help the patients perform their daily activities.
Keywords: Retinal dystrophies; Pigmentary retinosis; Bardet-Biedl syndrome.
INTRODUCTION
Bardet-Biedl syndrome (BBS) is a rare autosomal recessive disease that was first described by Laurence-Moon in 1866 and additional cases were described by Bardet and Biedl between 1920 and 1922 in the British Journal of Ophthalmology 1. The current prevalence in the population ranges from 1:140.000 to 160.000 live births 2. The main clinical manifestations are retinal dystrophy, polydactyly, obesity, mental retardation, and hypogenitalism 3. Among these symptoms, retinal dystrophy is the most consistent finding and causes progressive visual loss from childhood.
CASE REPORT
The WBR patient, a 21-year-old male student, brown, seeked care for follow-up of pigmentary retinosis (PR) diagnosed in childhood. According to the companion, the patient had cognitive and auditory deficits since birth and bilateral polydactyly in both hands. Furthermore, his family’s medical history indicates that he has a brother with the same diagnosis. The physical examination revealed obesity and the correction scars related to the treatment of hexadactilia of both hands (Figures 1A and 1B). In addition, the patient had previously been diagnosed with hypogonadism.
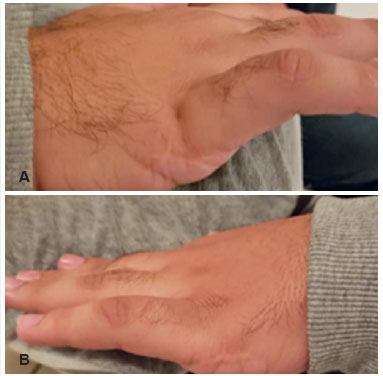
When performing the ophthalmological examination, count finger visual acuity, without correction, and the Snellen chart were used to evaluate both eyes. Intraocular pressure and biomicroscopy were normal. When examining the fundus of the eye, pale optic disc, narrowing of vessels, atrophy of retinal pigment epithelium (RPE) and pigmentary changes with the appearance of bone spicules on the periphery of both eyes were presented. Visual field examination proved to be unreliable in both eyes.
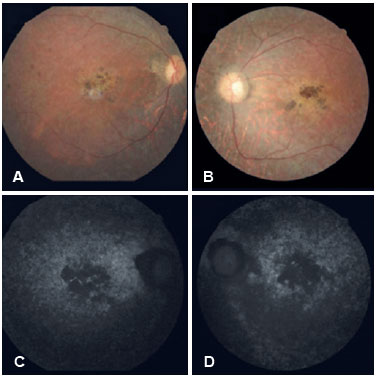
In both eyes, retinography (Figures 2A and 2B) revealed the presence of transparent media, pale papilla, physiological cupping, arteriolar thinning, pigmentary changes with the appearance of bone spicules on the periphery, and atrophy in the macular region with probable pigment mobilization. We used autofluorescence (Figures 2C and 2D) to identify RPE atrophy indicated by the presence of hypoautofluorescent images in the macular region in both eyes. It was concluded that in addition to PR, the patient’s diagnosis fits the BBS criteria.
DISCUSSION
BBS is a rare autosomal recessive disorder with clinical and genetic heterogeneity. Stigglebout et al. described the criteria for the diagnosis of BBS as the presence of four main characteristics or the combination of three primary and two secondary characteristics. The main characteristics include retinal dystrophy, polydactyly, obesity, learning difficulty, hypogonadism, and renal anomalies. Secondary characteristics include speech disorders, strabismus, cataract, astigmatism, syndactyly, brachydactyly, developmental delay, polyuria, polydipsia, small tooth root, hypodontia, high palate, left ventricular hypotrophy, diabetes mellitus, congenital heart disease, hepatic fibrosis, ataxia, poor coordination, and imbalance 4-6.
The BBS etiology is uncertain and with only a few cases described in the literature 7. The BBS mutation spectrum is divergent between populations; notably, in some patients, the disease can only manifest in the presence of three mutations 2.
Although BBS was first described more than 80 years ago 8, by 2015, researchers have identified the 19 causative genes (BBS1-19) that are associated with the condition, and mutations in these genes can explain the occurrence of BBS in >80% of the patients. The superimposed phenotypes between the ciliopathies, in addition to the high intra-family and inter-family variability in the clinical presentation, further complicate the diagnosis of this syndrome 9.
Patients with this syndrome have dystrophy with PR phenotype and the diagnosis is usually confirmed during childhood when visual problems caused by PR are identified. PR is not a disease but a group of diseases caused by numerous genetic mutations and is responsible for the gradual degeneration of light-sensitive retinal cells. Degeneration of retinal cones and rods results in night blindness, the first symptom of PR, which makes it difficult for the patient see in low levels of light. Subsequently, PR is responsible for the progressive loss of peripheral vision, and the loss of central vision may occur during childhood or adolescence 7.
Over time, the patient’s vision continues to become more restricted. Eventually, the cones are affected with the progression of the disease, consequently resulting in decreased visual acuity, perception of colors and details, and central vision 10,11.
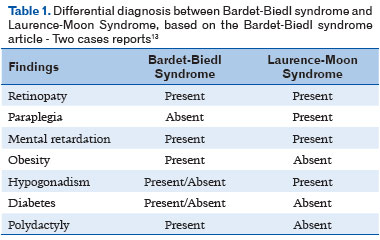
BBS is often confused with Laurence-Moon syndrome (LMS) and vice versa owing to the similarities between the syndromes 12. Contrary to BBS, individuals with LMS almost always have neurological problems, they and rarely present with polydactyly, a defining characteristic of BBS (Table 1). LMS is extremely rare, so few cases have been documented.
Although there is currently no treatment for BBS, early diagnosis is important for possible genetic counseling and prenatal care to ensure regular follow-up examinations and interventions for the child through routine weight assessment, blood pressure measurement, ophthalmological examinations, renal imaging studies, and psychological support. It is recommended that all patients diagnosed with BBS syndrome should be accompanied by a multidisciplinary team. Currently, INN-Voretigene neparvovec is indicated for the treatment of patients with loss of vision due to hereditary retinal dystrophy caused by confirmed RPE65 gene mutations and for those who have insufficient viable retinal cells 14.
BBS is a rare autosomal recessive disease and the diagnosis is usually confirmed in childhood, which did not occur in this specific case. The patient had the following main clinical characteristics: retinal dystrophy, polydactyly, obesity, male hypogonadism, and cognitive impairment (Table 1). In conclusion, the diagnosis for this syndrome is often characterized by retinal dystrophy, considering the fact that it is present in nearly 90% of the cases previously reported. Although there is currently no treatment for BBS, early diagnosis is important for multidisciplinary follow-up. The ophthalmologist plays an important role, and the patient should undergo periodic consultations.
REFERENCES
1. O’mahony PF. Síndrome de Laurence-Moon-Biedl. The New England Journal of Medicine, Londres, 1954 setembro. [acesso em: 2019 ago. 07]; 439-440. Disponível em: https://www.nejm.org/doi/full/10.1056/NEJM195409092511107
2. M’hamdi O, Ouertani I, Chaabouni-Bouhamed H. Update on the genetics of bardet-biedl syndrome. Molecular Syndromology, Londres. 2014. [acesso em: 2019 ago. 07]; 5(2):51-6. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/24715851
3. Toledo NB et al. Síndrome de Bardet-Biedl: série de caso e revisão de literatura. Revista Brasileira de Oftalmologia, Rio de Janeiro. 2018 novembro-dezembro. [acesso em: 2019 ago. 12]; 77(6.) Disponível em: http://www.scielo.br/pdf/rbof/v77n6/0034-7280-rbof-77-06-0360.pdf
4. Lo KT, Remulla J, Santiago AP. Manifestations of Bardet-Biedl syndrome. Philipp J Ophthalmol. 2004. [acesso em: 2019 ago. 10];29(2):94-8. Disponível em: https://apamedcentral.org/search.php?where=aview&id=10.0000/pjo.2004.29.2.94&code=0014PJO&vmode=PUBREADER
5. Siopa L, Grego M, Cossa J, Pinguinha A. [Bardet-Biedl Syndrome]. Acta Med Port. 2002. [acesso em 2019 ago. 10]; 15(1):51-4. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/12025454
6. Stigglebout W. The Bardet-Biedl syndrome: including Hutchinson- LaurenceMoon syndrome. In: Vinkin PJ, Bruyn GW, editors. Neuroretinal degenerations: Handbook of clinical neurology. Amsterdam: North-Holland; 1972. [acesso em: 2019 ago. 10]; 380-412. Disponível em: https://jamanetwork.com/journals/jamaophthalmology/fullarticle/634082
7. Sahel JÁ, Marazova K, Audo I. Clinical characteristics and current therapies for inherited retinal degenerations. Cold Spring harbor Perspectives in Medicine, Bethesd. 2014 agosto. [acesso em: 2019 set. 06]. Disponível em: http://perspectivesinmedicine.cshlp.org/content/5/2/a017111
8. Sánchez SC. et al. Exploring genotype-phenotype relationships in Bardet-Biedl syndrome families. Journal of Medical Genetics, Londres. 2015 agosto. [acesso em: 2019 ago. 10]; 503-13. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/26082521
9. Biesecker LGUS. National Library of Medicine, Bethesda. 2019 junho. [acesso em: 2019 ago. 10]. Disponível em: https://clinicaltrials.gov/ct2/show/NCT00078091?cond=Bardet-Biedl&rank=2
10. FOUNDATION FIGHTING BLINDNESS. Bardet-Biedl syndrome (BBS). Foundation Fighting Blindness, Columbia. [s/d]. [acesso em: 2019 ago. 10]. Disponível em: https://www.fightingblindness.org/diseases/bardet-biedl-syndrome-bbs#research
11. FOUNDATION FIGHTING BLINDNESS. Retinits Pigmentosa. [s/d]. [acesso em: 2019 ago. 10]. Disponível em: https://www.fightingblindness.org/diseases/retinitis-pigmentosa
12. RETINA BRASIL. Doenças: Síndrome de Bardet-Biedl. Retina Brasil. [s/d]. [acesso em: 2019 ago. 10]. Disponivel em: http://retinabrasil.org.br/site/doencas/sindrome-de-laurence-moon-bardet-biedl
13. Lavinsky J, Goldhardt R, Ariente S, Domingues C et al. Bardet-Biedl syndrome - Two case reports. Arq Bras Oftalmol. 2003. [acesso em: 2019 ago. 10]; 66:675-80. Disponível em: http://www.scielo.br/pdf/abo/v66n5/18160.
14. Dias M. et al. Molecular genetics and emerging therapies for retinitis pigmentosa: Basic research and clinical perspectives. Elsevier. 2017. [acesso em: 2019 ago. 10].
AUTHOR’S INFORMATION





Financiamento: Declaram não haver.
Conflitos de Interesse: Declaram não haver.
Recebido em:
1 de Novembro de 2019.
Aceito em:
17 de Junho de 2020.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados

 Ler em inglês
Ler em inglês
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket