

Carlos Augusto Moreira Neto1; Letícia Costa Almeida Mariuzzo1; Vera Lúcia Rodrigues Falcão1; José Fábio de Oliveira Miranda1; Paula Maria Haggi Fregadolli1; Lunna Pires Moreira2
DOI: 10.17545/eOftalmo/2020.0018
ABSTRACT
Bardet-Biedl syndrome (BBS) is a rare autosomal recessive disease that is typically characterized by the following symptoms: polydactyly, obesity, mental retardation, hypogenitalism, and retinal dystrophy. Despite the absence of some of the abovementioned symptoms, retinal dystrophy is consistently observed in BBS patients. WBR, a 21-year-old man, seeked care for follow-up of pigmentary retinosis that was diagnosed since childhood. He has a brother with the same diagnosis. Count finger visual acuity was used to evaluate both eyes. Fundus examination revealed a pale optic disc, narrowing of vessels, atrophy of retinal pigment epithelium, and pigmentary changes with the appearance of bone spicules on the periphery. Physical examination revealed the correction scars related to the treatment of hexadactilia of both hands, along with other symptoms such as obesity, auditory, and cognitive deficits. In addition, he had previously been diagnosed with hypogonadism. Computerized campimetry proved to be unreliable in both eyes. It was observed that in addition to pigmentary retinosis, the patient met the diagnostic criteria of BBS. BBS is a rare disease with a currently unknown etiology, and there have only been a few cases of this condition have been reported in the literature. Although there is currently no treatment, early diagnosis is important for multidisciplinary follow-up to help the patients perform their daily activities.
Keywords: Retinal dystrophies; Pigmentary retinosis; Bardet-Biedl syndrome.
RESUMO
A Síndrome de Bardet-Biedl é uma rara doença autossômica recessiva caracterizada por: polidactilia, obesidade, retardo mental, hipogenitalismo e distrofia retiniana. Alguns dos achados acima podem estar ausentes, mas a distrofia retiniana é achado consistente.WBR, 21 anos, masculino, procura atendimento para acompanhamento de retinite pigmentosa diagnosticado desde a infância. Possui irmão com o mesmo diagnóstico. Acuidade visual de conta dedos em ambos os olhos. Ao exame de fundo de olho apresenta disco óptico pálido, estreitamento de vasos, atrofia de epitélio pigmentar da retina e alterações pigmentares com aspecto de espículas ósseas na periferia. Ao exame físico, apresenta cicatrizes de correção da hexadactilia de mãos, obesidade, déficit auditivo e cognitivo. Além disso, possui diagnóstico prévio de hipogonadismo. Campimetria computadorizada mostrou-se não confiável em ambos os olhos. Observou-se então, que além de retinite pigmentosa o paciente se enquadrava nos critérios de Síndrome de Bardet-Biedl. A síndrome de Bardet-Biedl é uma rara doença de etiologia ainda incerta, com apenas poucos casos descritos na literatura. Embora atualmente não haja tratamento, o diagnóstico precoce é importante para o acompanhamento multidisciplinar, melhorando o desempenho em suas atividades diárias.
Palavras-chave: Distrofias retinianas; Retinose pigmentar; Síndrome de Bardet-Biedl.
INTRODUÇÃO
A Síndrome de Bardet-Biedl (SBB) é uma rara doença autossômica recessiva que foi descrita pela primeira vez por Laurence-Moon em 1866 e casos adicionais foram descritos por Bardet 1920 e Biedl 1922 no British Journal of Ophthalmology em 1866 1. A prevalência atual na população varia de 1:140.000 – 160.000 nascidos vivos 2. As principais manifestações clínicas são distrofia retiniana, polidactilia, obesidade, retardo mental e hipogenitalismo 3. A distrofia retiniana é o achado mais consistente e causa progressiva perda visual a partir da infância.
RELATO DO CASO
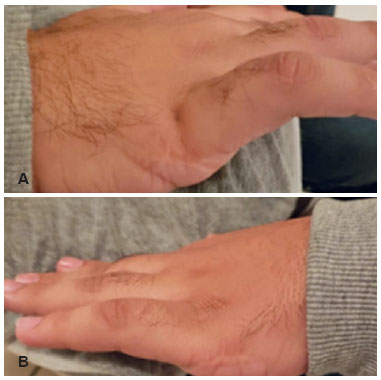
O paciente WBR, 21 anos, masculino, pardo, estudante, procura atendimento para acompanhamento de retinite pigmentosa (RP) diagnosticado na infância. Segundo a acompanhante, o paciente apresentava déficit cognitivo e auditivo desde o nascimento e polidactilia bilateral em mãos. Em sua história familiar possui irmão com o mesmo diagnóstico. Ao exame físico apresentava obesidade, cicatrizes de correção da hexadactilia de mãos (Figuras 1A e 1B) e possui diagnóstico prévio de hipogonadismo.
Na realização do exame oftalmológico, a acuidade visual,sem correção, era de conta dedos em ambos os olhos (AO), utilizando a tabela de Snellen. A pressão intraocular e a biomicroscopia apresentavam-se normais. Ao exame de fundo de olho apresenta disco óptico pálido, estreitamento de vasos, atrofia de epitélio pigmentar da retina e espíiculas em periferia AO. O exame de campo visual mostrou-se não confiável em ambos os olhos.
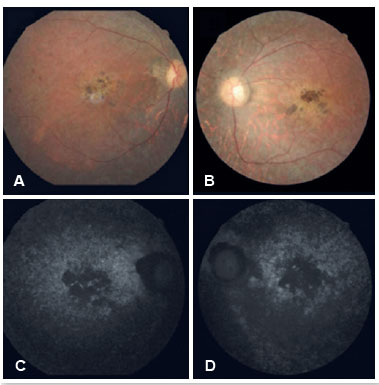
Na retinografia (Figuras 2A e 2B) evidenciou-se, em ambos os olhos, meios transparentes, papila com palidez, escavação fisiológica, afinamento arteriolar, presença de alterações pigmentares com aspecto de espículas ósseas na periferia e presença de atrofia em região macular com provável mobilização de pigmento. À autofluorescência (Figuras 2C e 2D), foi observada, em ambos os olhos, a presença de imagens hipoautofluorescentes em região macular, indicando atrofia de EPR. Concluiu-se então, que além de RP o paciente se enquadra nos critérios de SBB.
DISCUSSÃO
A Síndrome de Bardet-Bield (SBB) é uma desordem autossômica recessiva rara, com heterogeneidade clínica e genética. Stigglebout et al. descreveram os critérios para o diagnóstico da SBB como: a presença de quatro caraterísticas principais ou a combinação de três características principais mais duas secundárias. As características principais são: distrofia retiniana, polidactilia, obesidade, dificuldade de aprendizado, hipogonadismo e anomalias renais. Dentre as características secundárias destacam-se: distúrbio da fala, estrabismo, catarata, astigmatismo, sindactilia, braquidactilia, atraso no desenvolvimento, poliúria, polidipsia, raiz dos dentes pequena, hipodontia, palato alto, hipotrofia do ventrículo esquerdo, diabetes mellitus, doença cardíaca congênita, fibrose hepática, ataxia, má coordenação e desequilíbrio (4-6).
A etiologia é incerta e com apenas poucos casos descritos na literatura 7. O espectro de mutação BBS é divergente entre as populações. Alguns pacientes necessitam de três mutações para manifestarem a doença 2.
Embora a SBB tenha sido descrita há mais de 80 anos 8, até 2015, 19 genes causadores (BBS1-19) foram envolvidos, cujas mutações explicariam mais de 80% dos pacientes. Os fenótipos sobrepostos entre as ciliopatias, além da alta variabilidade intrafamiliar e interfamiliar na apresentação clínica, complicam ainda mais o diagnóstico dessa síndrome 9.
Indivíduos com esta síndrome têm distrofia com fenótipo de retinose pigmentar (RP) e o diagnóstico geralmente é confirmado na infância, quando problemas visuais devido à RP são descobertos. A RP não constitui uma doença e sim um grupo de doenças, causadas por inúmeras mutações genéticas, cujo traço comum é a degeneração gradativa das células da retina sensíveis à luz. Com a degeneração dos cones e bastonetes da retina, o primeiro sintoma da RP é cegueira noturna, que faz com que o indivíduo tenha dificuldade de enxergar com baixos níveis de luz, em seguida a RP provoca a perda progressiva da visão periférica, e a perda da visão central pode ocorrer durante a infância ou adolescência 7.
A visão se torna mais restrita ao longo do tempo. Se e quando a doença progride e os cones são afetados, a acuidade visual, a percepção de cores e detalhes, e a visão central diminuem 10,11.
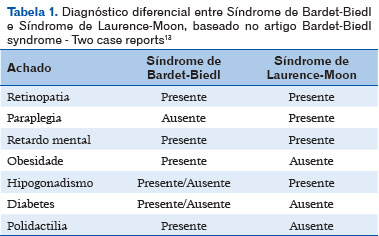
A SBB é muitas vezes confundida com síndrome de Laurence-Moon (SLM). Indivíduos com SLM quase sempre possuem problemas neurológicos, mas raramente apresentam polidactilia, que é uma característica definidora de SBB, enquanto que problemas neurológicos quase nunca ocorrem (Tabela 1). A SLM é extremamente rara, poucos casos foram documentados. Devido à semelhança das síndromes, Bardet-Biedl é muitas vezes referida como Laurence-Moon e vice versa 12.
Embora atualmente não exista tratamento para a SBB, o diagnóstico precoce é importante para um possível aconselhamento genético e durante o pré-natal, para orientar o acompanhamento da criança através de uma avaliação regular do peso, da pressão arterial, exames oftalmológicos, estudos de imagem renais e apoio psicológico. Recomenda-se que todos os pacientes com diagnóstico de síndrome de SBB sejam acompanhados por uma equipe multidisciplinar. 9 Até o presente momento, o INN-voretigene neparvovec é indicado no tratamento de doentes com perda de visão devido a distrofia retiniana hereditária causada por mutações no gene RPE65 confirmadas e que tenham suficientes células retinianas viáveis 14.
A SBB é uma rara doença autossômica recessiva e o diagnóstico normalmente é confirmado na infância, o que não ocorreu nesse caso. O paciente apresentava as principais características clinicas: distrofia retiniana, polidactilia, obesidade, hipogonadismo masculino e comprometimento cognitivo (Tabela 1). Concluindo-se então, o diagnóstico presumível para a síndrome relatada, sendo a distrofia retiniana muitas vezes o que caracteriza o diagnóstico, por estar presente em cerca de 90% dos casos já descritos. Embora atualmente não haja tratamento para a SBB, o diagnóstico precoce é importante para um acompanhamento multidisciplinar. O oftalmologista desempenha um importante papel e o paciente deve realizar consultas periódicas.
REFERÊNCIAS
1. O’mahony PF. Síndrome de Laurence-Moon-Biedl. The New England Journal of Medicine, Londres, 1954 setembro. [acesso em: 2019 ago. 07]; 439-440. Disponível em: https://www.nejm.org/doi/full/10.1056/NEJM195409092511107
2. M’hamdi O, Ouertani I, Chaabouni-Bouhamed H. Update on the genetics of bardet-biedl syndrome. Molecular Syndromology, Londres. 2014. [acesso em: 2019 ago. 07]; 5(2):51-6. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/24715851
3. Toledo NB et al. Síndrome de Bardet-Biedl: série de caso e revisão de literatura. Revista Brasileira de Oftalmologia, Rio de Janeiro. 2018 novemb ro-dezembro. [acesso em: 2019 ago. 12]; 77(6.) Disponível em: http://www.scielo.br/pdf/rbof/v77n6/0034-7280-rbof-77-06-0360.pdf
4. Lo KT, Remulla J, Santiago AP. Manifestations of Bardet-Biedl syndrome. Philipp J Ophthalmol. 2004. [acesso em: 2019 ago. 10];29(2):94-8. Disponível em: https://apamedcentral.org/search.php?where=aview&id=10.0000/pjo.2004.29.2.94&code=0014PJO&vmode=PUBREADER
5. Siopa L, Grego M, Cossa J, Pinguinha A. [Bardet-Biedl Syndrome]. Acta Med Port. 2002. [acesso em 2019 ago. 10]; 15(1):51-4. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/12025454
6. Stigglebout W. The Bardet-Biedl syndrome: including Hutchinson- LaurenceMoon syndrome. In: Vinkin PJ, Bruyn GW, editors. Neuroretinal degenerations: Handbook of clinical neurology. Amsterdam: North-Holland; 1972. [acesso em: 2019 ago. 10]; 380-412. Disponível em: https://jamanetwork.com/journals/jamaophthalmology/fullarticle/634082
7. Sahel JÁ, Marazova K, Audo I. Clinical characteristics and current therapies for inherited retinal degenerations. Cold Spring harbor Perspectives in Medicine, Bethesd. 2014 agosto. [acesso em: 2019 set. 06]. Disponível em: http://perspectivesinmedicine.cshlp.org/content/5/2/a017111
8. Sánchez SC. et al. Exploring genotype-phenotype relationships in Bardet-Biedl syndrome families. Journal of Medical Genetics, Londres. 2015 agosto. [acesso em: 2019 ago. 10]; 503-13. Disponível em: https://www.ncbi.nlm.nih.gov/pubmed/26082521
9. Biesecker LGUS. National Library of Medicine, Bethesda. 2019 junho. [acesso em: 2019 ago. 10]. Disponível em: https://clinicaltrials.gov/ct2/show/NCT00078091?cond=Bardet-Biedl&rank=2
10. FOUNDATION FIGHTING BLINDNESS. Bardet-Biedl syndrome (BBS). Foundation Fighting Blindness, Columbia. [s/d]. [acesso em: 2019 ago. 10]. Disponível em: https://www.fightingblindness.org/diseases/bardet-biedl-syndrome-bbs#research
11. FOUNDATION FIGHTING BLINDNESS. Retinits Pigmentosa. [s/d]. [acesso em: 2019 ago. 10]. Disponível em: https://www.fightingblindness.org/diseases/retinitis-pigmentosa
12. RETINA BRASIL. Doenças: Síndrome de Bardet-Biedl. Retina Brasil. [s/d]. [acesso em: 2019 ago. 10]. Disponivel em: http://retinabrasil.org.br/site/doencas/sindrome-de-laurence-moon-bardet-biedl
13. Lavinsky J, Goldhardt R, Ariente S, Domingues C et al. Bardet-Biedl syndrome - Two case reports. Arq Bras Oftalmol. 2003. [acesso em: 2019 ago. 10]; 66:675-80. Disponível em: http://www.scielo.br/pdf/abo/v66n5/18160.
14. Dias M. et al. Molecular genetics and emerging therapies for retinitis pigmentosa: Basic research and clinical perspectives. Elsevier. 2017. [acesso em: 2019 ago. 10].
INFORMAÇÃO DOS AUTORES





Funding source: The author declares there was none.
Conflict of interest: The author declares there is none.
Received on:
November 1, 2019.
Accepted on:
June 17, 2020.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados

 Ler em português
Ler em português
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket