

Victor Fellipe Justiniano Barbosa1; Thiago Oliveira Ferreira2; Alex Pereira Ramos3; Marlon Mohamud Vilagra4; Cáritas Antunes Lacerda5
DOI: 10.17545/eoftalmo/2017.111
ABSTRACT
Kearns–Sayre syndrome (KSS) is primarily caused by changes in mitochondrial deoxyribonucleic acid (mtDNA). The standard diagnostic criteria for KSS are progressive external ophthalmoplegia, pigmentary retinopathy, and cardiac conduction disorders. This report describes the case of a patient who sought medical treatment primarily because of unilateral ptosis at 10 years of age. He was diagnosed with KSS at 19 years of age. At 28 years of age, the presence of third-degree atrioventricular block was detected, and he underwent cardiac pacemaker implantation. KSS is a rare disease; therefore, physicians should strongly suspect the disease based on the symptoms.
Keywords: Neoplasms; Heart Diseases; Blefaroptosis; Ophthalmoplegia; Retinitis Pigmentosa.
RESUMO
A síndrome de Kearns-Sayre (SKS) tem como fatores causais alterações no DNA mitocondrial (mtDNA), e tem como critérios diagnósticos mais aceitos a tríade: oftalmoplegia externa progressiva, retinopatia pigmentar e distúrbios de condução cardíaca. Neste relato, descreve-se o caso de paciente que apresentou como queixa principal inicial um quadro de ptose palpebral unilateral aos 10 anos de idade. Foi diagnosticado com SKS aos 19 anos de idade e, aos 28 anos de idade, foi detectada a presença de um bloqueio cardíaco atrioventricular total e realizou implantação imediata de marca-passo cardíaco. A SKS é uma doença rara, portanto o médico deve possuir grande suspeição clínica para o seu diagnóstico.
Palavras-chave: Cardiopatias; Blefaroptose; Oftalmoplegia; Retinite Pigmentosa.
RESUMEN
El síndrome de Kearns-Sayre (SKS) tiene como factores causales alteraciones en el ADN mitocondrial (mtDNA). Tiene como criterios diagnósticos más aceptados la tríada: oftalmoplejía externa progresiva, retinopatía pigmentar y disturbios de conducción cardíaca. En este artículo, se describe el caso de un paciente que presentó como queja principal inicial un cuadro de ptosis palpebral unilateral a los 10 años de edad. Fue diagnosticado con SKS a los 19 años de edad. A los 28 años de edad, se detectó la presencia de un bloqueo cardíaco atrioventricular total y se realizó implantación inmediata de marcapaso cardíaco. El SKS es una enfermedad rara, por lo tanto, el médico debe tener un gran factor de sospecha c línica para su diagnóstico.
Palabras-clave: Cardiopatías; Blefaroptosis; Oftalmoplejía; Retinitis Pigmentosa.
INTRODUCTION
In 1958, Kearns and Sayre wrote a report of two cases of a syndrome characterized by external ophthalmoplegia, pigmentary retinopathy, and cardiac conduction disorders. However, this condition was recognized as a syndrome only in 1965 and was renamed as Kearns–Sayre syndrome (KSS).1
KSS is caused by changes in mitochondrial DNA (mtDNA); the most commonly observed deletions range from 1.3 to 8.0 kilobases in size. KSS leads to clinical manifestations, including upper and lower limb weakness, fatigue, external ophthalmoplegia, ptosis, and low visual acuity.1 This syndrome is rare, with an incidence of 1.6 cases for every 100,000 individuals.2
Clinical manifestations usually begin before 20 years of age. Patients’ ophthalmologic assessment usually reveals progressive external ophthalmoplegia, ptosis, retinal degeneration, pigmented epithelium, and symptomatic cardiac block. Short stature, high CSF protein level, proximal myopathy, and cerebellar dysfunction signs, including ataxia, deafness, endocrinopathy, and acid–base imbalances, as well as other multisystem manifestations, are also common.3
Etiologic investigation is imperative because many patients develop sudden heart blocks as well as dementia and balance disorders.
Cardiac conduction disorders cause high mortality and occur in approximately 57% of cases.1,3
The primary objective of this study was to describe the clinical case of a patient with KSS and analyze the clinical aspects of the case to improve early diagnoses and differential diagnoses in cases of similar clinical conditions. The secondary objective was to demonstrate the importance of the etiological investigation of clinical ophthalmologic conditions because they may be associated with diseases with severe systemic complications, as in the case reported herein.
CASE REPORT
A 36-year-old man presented with chronic myopathy, which primarily affected the extraocular muscles, and the first symptoms appeared at 10 years of age. The patient reported that ptosis began unilaterally (on the left side) at 10 years of age and developed into bilateral ptosis within 1 year. He was diagnosed with KSS in 1998 inSanta Casa de Misericórdia Hospital, Rio de Janeiro, Brazil, after the exclusion of important differential diagnosis. Differential diagnosis included oculopharyngeal muscular dystrophy (which commonly begins at 50 years of age and is characterized by severe dysphagia and a myopathic pattern in biopsy) and myasthenia gravis, which was excluded because the results of electroneuromyography and acetylcholine receptor antibody test were normal. Computed tomography and nuclear magnetic resonance imaging were performed to discard possible macroscopic structural damage such as tumors. Cerebrospinal fluid was also analyzed, and the results were confirmed by muscular biopsy and histopathological examination at the Department of Pathology of the Clementino Fraga Filho University Hospital of the Federal University of Rio de Janeiro. In histological sections stained with hematoxylin and eosin, most of the muscle fibers were preserved and a slightly irregular size was observed. Gömöri trichrome staining revealed the presence of fibers with a small degree of atrophy and/or increased reddish granular material. These fibers were more evident in a succinate dehydrogenase assay and exhibited increased and peripheral oxidative activity. Fat accumulation was also observed in these fibers, but an increase in glycogen was not detected in the sections stained with periodic acid–Schiff. The results of ATPase assay indicated a mosaic distribution pattern of the fibers. These changes were designated as mitochondriopathy compatible with KSS. The patient was then administered coenzyme Q10 (CoQ10).
After 1 year, the patient developed complete paralysis of the extraocular muscles, a critical functional limitation, and 19 years of age, he underwent surgical correction of ptosis (Figures 1 and 2). The ocular diameter of the palpebral fissure was increased from 5 mm to 9 mm in the right eye and from 4 mm to 8 mm in the left eye postoperatively.


During the same period, the patient experienced a change in the tone of his voice (dysphonia), fatigue, dyspnea upon exertion, and hearing loss. Later, he reported recurrent episodes of dyspnea upon moderate exertion, as well as syncope, intense fatigue, and headache. In 2008, he was diagnosed with primary open-angle glaucoma and pigmentary dispersion syndrome.
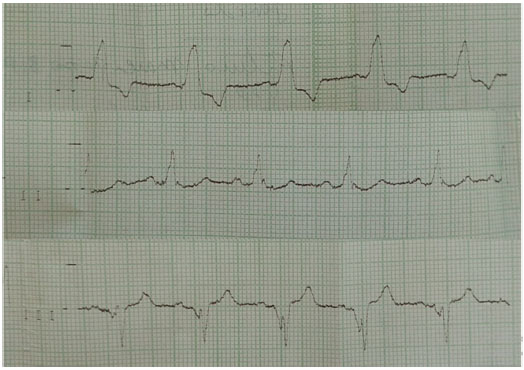
At 28 years of age, cardiac examination with 24 hour holter, was requested after the occurrence of dyspnea crisis following moderate exertion and an episode of syncope. The test results indicated the presence of cardiac conduction disorder and third-degree atrioventricular block, with a heart rate of 33–43 bpm. Therefore, the patient underwent permanent pacemaker implantation (Figures 3 and 4).

During the clinical examination, the patient was cooperative, hydrated, and aware of time and space. Blood circulation and skin tone were normal, and his vital signs were stable. The patient had a heart rate of 80 bpm, respiratory rate of 16 breaths per minute, and systemic arterial pressure of 110/70 mmHg. Cardiovascular examination revealed a palpable ictus approximately two fingerbreadths in length located on the left midclavicular line in the fifth intercostal space, absence of visible pulsation in the pericardium, cardiac auscultation with regular heart rhythm, normal heart sounds, and presence of a systolic murmur in the mitral area. The respiratory system exhibited no deformities or signs of excess respiratory effort; tactile fremitus was preserved, bilateral vesicular murmur was audible, adventitious noises were absent, and chest expansion was not impaired. The patient’s abdomen was flaccid and peristaltic, without pain upon superficial or deep palpation, and there were no palpable masses or visceromegaly. The patient’s upper and lower limbs had symmetrically palpable pulse, preserved reflexes, and no visible edema. The findings of sensory analysis indicated the presence of bilateral ptosis, dysphonia (hypernasal speech), and auditory profile compatible with mild-to-moderate sensorineural loss on the left side and light-to-moderate sensorineural loss on the right side; this diagnosis was confirmed using tympanometry. Ophthalmologic examination revealed a visual acuity of 20/25 in the right eye (RE) and 20/60 in the left eye (LE). Anterior segment biomicroscopy in RE and LE showed clear conjunctivae, transparent cornea with a mild inferior superficial punctate keratitis, deep anterior chamber, no signs of inflammation, regular and normal colored pupils, and transparent lenses. The patient had significantly limited extrinsic muscle mobility based on the examination of ductions and versions and absence of Bell’s reflex. Fundoscopy of RE and LE exhibited strong dispersion of the retinal pigment epithelium and areas of retinal atrophy, optic disc with free borders, and cupping of 0.9 ´ 0.9, with the presence of elongated pores in the lamina cribrosa in RE and cupping of 0.8 ´ 0.8 in LE; reduced arteriolar calibers and normal paths. Ophthalmic polyacrylic acid and carboxymethylcellulose ophthalmic eye drops were prescribed.
DISCUSSION
The diagnosis of KSS is clinical. Diagnosis is suspected after early signs of the disease (before 20 years of age), including ptosis, ophthalmoplegia, and pigmentary retinopathy. Approximately 57% of patients with KSS have signs of cardiac dysfunction. Therefore, cardiac monitoring is critical because of the associated risk of atrioventricular block and sudden death.
Mitochondrial dysfunction due to spontaneous mutations is a causal factor of KSS and leads to changes in mtDNA. Although it has long been known that mtDNA is transmitted from the mother to the fetus, the underlying mechanism of transmission has been recently clarified. In contrast to a large number of mitochondria in oocytes, spermatozoa contain a few mitochondria, and these are not transmitted to the offspring. mtDNA is thus inherited from the mother and not the father.4
In these cases, most mutations, including germ-cell mutations, are mtDNA deletions that sporadically occur before embryogenesis and at the onset of embryogenesis.4 The deletions vary from 1.3 kb to 8 kb in size and occur at different positions in the mitochondrial genome. Two-thirds of the deletions occur between positions 8469 and 13147 of the genome, and the size of most deletions is 4.9 kb. Despite the variability in size, these deletions produce similar phenotypes.4
The need for biopsy is controversial,5 although it is necessary for diagnostic confirmation by allowing the visualization of significant changes in muscle fibers. Other noticeable changes include the presence of Gömöri spots between the fibers and a large number of mitochondria on light microscopy. In this case, there is a positive correlation between the number of mitochondria detected in the biopsy and the probability of mutations.6
Dark red-stained muscle fibers may also be observed and indicate mitochondrial disease. Other analyses may be performed on the sample, including immunohistochemical tests (which are more specific for analyzing the mitochondrial enzyme content), histochemical analysis of muscle tissue (such as those assessing enzyme activity of the electron transport chain), and mitochondrial DNA analysis.6
Because of the multisystemic nature of KSS,3,4 the search for correlations should be extensive. For instance, a significant number of abnormalities in the cardiovascular system may be correlated with KSS. Evidence of these correlations was found by Khambatta et al. in 35 patients with KSS who exhibited cardiovascular conditions.8 Six patients (17%) presented with syncope and four (11%) were lost because of sudden cardiac death. Further examinations suggested the presence of several cardiac abnormalities for which the use of a pacemaker could be indicated as a prophylactic treatment, including branch block [detected in 11 patients (31%)] and delayed conduction [detected in 23 patients (66%)]. The patient in our case report had third-degree atrioventricular block, and this condition was found in 31% of the patients in the study by Khambatta et al. For such patients, pacemakers were indicated.
KSS has no definitive treatment, and current options are palliative. CoQ10 is the most common choice of therapy. CoQ10 has been widely studied since its discovery in 1957. This component of the electron transport chain is involved in cellular aerobic respiration for generating energy in the form of adenosine triphosphate. The antioxidant and pro-oxidant properties of CoQ10 suggest that it plays an important role in the modulation of the cellular redox state in physiological and pathological conditions and may also play a role in the aging process. In several animal models of neurodegenerative diseases, CoQ10 was found to reduce disease progression, although the effects were temporary.7
However, there are disagreements over the efficacy of CoQ10. A randomized, prospective, double-blind study of 15 patients with KSS and chronic progressive external ophthalmoplegia compared CoQ10 to placebo and found no significant improvements in muscle dysfunction in a small group of patients with the syndrome.8
Atrioventricular block is often preceded by intraventricular conduction disturbances, such as left bundle branch block (LBBR) or isolated LBBR. The presence of third-degree atrioventricular block is an important prognostic factor in this syndrome. In a review by Berenberg et al. including five new cases and 30 cases from the literature, third-degree atrioventricular block was the cause of death in 20% of the cases.9
Most recent studies reported clinical improvements using CoQ10. In this case report, the patient reported a significant decrease in muscle fatigue, and CoQ10 produced a slight improvement in ophthalmoplegia. However, more evidence is needed to improve treatment and provide a better quality of life for patients with KSS.
CONCLUSION
KSS is a rare disease; therefore, physicians should have a strong clinical suspicion of the disease based on the symptoms. Physicians should be aware of the most common clinical manifestations and associated signs and symptoms to provide early diagnosis, thereby reducing the risks inherent to disease progression. Cardiovascular impairment with third-degree atrioventricular block causes high mortality. Further studies are needed to improve treatment and prevent complications. It is important to note that KSS is a complex disorder and therefore requires the involvement of various medical specialties, including neurology, cardiology, ophthalmology, and endocrinology. Therefore, patients need to receive regular follow-up care from these specialists because the prognosis is poor and the disorder is usually progressive.
REFERENCES
1. De Castro Junior H, Pena FM, Ribeiro ML, Martins WA. Síndrome de Kearns-Sayre: Relato de caso. Insuf Card. 2011;6(2):92-5.
2. Nasseh IE, Tengan CH, Kiyomoto BH, Gabbai AA. Doenças mitocondriais. Rev Neurociênc. 2001;9(2):60-9.
3. von Blotzheim SG, Borruat, FX; Hirt L. Myopathies mitochondriales Oculaires: un éventail de présentations cliniques. Klin Monatsbl Augenheilkd. 1998;212(5):299-300.
4. Ribeiro EM, Vasconcelos DO. Mitochondrial diseases. Rev Bras Med. 2006;63(4):169-73.
5. Zierz S, Jahns G, Jerusalem F. Coenzyme Q in serum and muscle of 5 patients with Kearns-Sayre syndrome and 12 patients with ophthalmoplegia plus. J Neurol. 1989;236(2):97-101. DOI: 10.1007/BF00314404
6. Rubin RM, Sadun AA. Ocular Myopathies. In: Yanoff M, Duker J. Ophthalmology. 3rd ed. Saint Louis: Mosby; 2008.
7. Santos GC, Antunes LMG, Santos AC, Bianchi MLP. Coenzyme Q10 and its effects in the treatment of neurodegenerative diseases. Braz J Pharm Sci. 2009;45(4):607-18. DOI: 10.1590/S1984-82502009000400002
8. Khambatta S, Nguyen DL, Beckman TJ, Wittich CM. Kearns-Sayre syndrome: a casa series of 35 adults and children. Int J Gen Med. 2014;7:325-32.
9. Marie SKN, Carvalho AAS, Fonseca LF, Carvalho MS, Reed UC, Scaff M. Kearns-Sayre syndrome "plus": classical clinical findings and dystonia. Arq Neuro-Psiquiatr. 1999;57(4):1017-23. DOI: 10.1590/S0004-282X1999000600020





Funding: No specific financial support was available for this study.
CEP Approval: Not applicable
Disclosure of potential conflicts of interest: None of the authors have any potential conflict of interest to disclose.
Received on:
August 8, 2017.
Accepted on:
December 6, 2017.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados
 Ler em português
Ler em português
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket