

Victor Fellipe Justiniano Barbosa1; Thiago Oliveira Ferreira2; Alex Pereira Ramos3; Marlon Mohamud Vilagra4; Cáritas Antunes Lacerda5
DOI: 10.17545/eoftalmo/2017.111
RESUMO
A síndrome de Kearns-Sayre (SKS) tem como fatores causais alterações no DNA mitocondrial (mtDNA), e tem como critérios diagnósticos mais aceitos a tríade: oftalmoplegia externa progressiva, retinopatia pigmentar e distúrbios de condução cardíaca. Neste relato, descreve-se o caso de paciente que apresentou como queixa principal inicial um quadro de ptose palpebral unilateral aos 10 anos de idade. Foi diagnosticado com SKS aos 19 anos de idade e, aos 28 anos de idade, foi detectada a presença de um bloqueio cardíaco atrioventricular total e realizou implantação imediata de marca-passo cardíaco. A SKS é uma doença rara, portanto o médico deve possuir grande suspeição clínica para o seu diagnóstico.
Palavras-chave: Cardiopatias; Blefaroptose; Oftalmoplegia; Retinite Pigmentosa.
ABSTRACT
Kearns–Sayre syndrome (KSS) is primarily caused by changes in mitochondrial deoxyribonucleic acid (mtDNA). The standard diagnostic criteria for KSS are progressive external ophthalmoplegia, pigmentary retinopathy, and cardiac conduction disorders. This report describes the case of a patient who sought medical treatment primarily because of unilateral ptosis at 10 years of age. He was diagnosed with KSS at 19 years of age. At 28 years of age, the presence of third-degree atrioventricular block was detected, and he underwent cardiac pacemaker implantation. KSS is a rare disease; therefore, physicians should strongly suspect the disease based on the symptoms.
Keywords: Neoplasms; Heart Diseases; Blefaroptosis; Ophthalmoplegia; Retinitis Pigmentosa.
RESUMEN
El síndrome de Kearns-Sayre (SKS) tiene como factores causales alteraciones en el ADN mitocondrial (mtDNA). Tiene como criterios diagnósticos más aceptados la tríada: oftalmoplejía externa progresiva, retinopatía pigmentar y disturbios de conducción cardíaca. En este artículo, se describe el caso de un paciente que presentó como queja principal inicial un cuadro de ptosis palpebral unilateral a los 10 años de edad. Fue diagnosticado con SKS a los 19 años de edad. A los 28 años de edad, se detectó la presencia de un bloqueo cardíaco atrioventricular total y se realizó implantación inmediata de marcapaso cardíaco. El SKS es una enfermedad rara, por lo tanto, el médico debe tener un gran factor de sospecha c línica para su diagnóstico.
Palabras-clave: Cardiopatías; Blefaroptosis; Oftalmoplejía; Retinitis Pigmentosa.
INTRODUÇÃO
Os autores Kearns e Sayre descreveram, em 1958, sob forma de relato de caso de dois pacientes, uma síndrome que cursava com oftalmoplegia externa, retinopatia pigmentar e distúrbio de condução cardíaco. Entretanto, somente em 1965 foi reconhecida como síndrome e passou a ser denominada síndrome de Kearns-Sayre (SKS).1
A SKS tem como fatores causais alterações no DNA mitocondrial (mtDNA), sendo mais comumente observadas deleções entre 1,3 a 8 kilobases do braço do mtDNA. Isso leva a manifestações clínicas de fraqueza muscular de membros superiores e inferiores, fadiga, oftalmoplegia externa, ptose palpebral e baixa da acuidade visual1. É considerada uma síndrome rara e, nesse sentido, estima-se que a proporção de casos seja de 1,6 para cada 100.000 indivíduos.2
As manifestações clínicas classicamente iniciam-se antes dos 20 anos de idade. Na avaliação oftalmológica dos pacientes, observa-se oftalmoplegia externa progressiva, ptose palpebral, degeneração da retina e epitélio pigmentado, bloqueio de condução cardíaco sintomático. Baixa estatura, hipoproteinorraquia, miopatia proximal, sinais de disfunção cerebelar como ataxia, surdez, endocrinopatias, e distúrbios do equilíbrio ácido básico são frequentemente associados, entre outras manifestações multissistêmicas.3
A investigação etiológica é imprescindível, haja vista que muitos pacientes desenvolvem bloqueio de condução cardíaco súbito, assim como demência e distúrbios de equilíbrio.
O distúrbio de condução cardíaco é responsável pela alta mortalidade da síndrome e ocorre em cerca de 57% dos casos.1,3
O objetivo deste estudo é descrever o caso clínico de um paciente com SKS e analisar os aspectos clínicos, a fim de contribuir para o seu diagnóstico precoce, bem como diagnóstico diferencial de quadros clínicos semelhantes. Além disso, demonstrar a importância da investigação etiológica dos quadros clínicos oftalmológicos, uma vez que podem estar associados a doenças com repercussões sistêmicas graves, como no caso retratado.
RELATO DE CASO
Homem, de 36 anos, apresenta miopatia crônica com predomínio de acometimento da musculatura extraocular, com primeiros sintomas iniciados aos 10 anos. Relata que o quadro de ptose palpebral se iniciou unilateralmente à esquerda aos 10 anos de idade, com evolução em um ano para ptose bilateral. Chegou-se, então, ao diagnóstico de síndrome de Kearns-Sayre em 1998 (Santa Casa de Misericórdia do Rio de Janeiro), após exclusão de diagnósticos diferenciais importantes, como a distrofia muscular óculo-faríngea, que comumente se inicia na quinta década de vida, cursa com intensa disfagia e tem, à biópsia, um padrão miopático; miastenia gravis, por meio da eletroneuromiografia normal e teste do anticorpo antirreceptor da acetilcolina negativo; realização tomografia computadorizada e ressonância nuclear magnética para visualização de possíveis danos estruturais macroscópicos, como tumores; e análise do líquor cefalorraquidiano, confirmado através de biópsia muscular e exame histopatológico (Serviço de Patologia do HUCFF-UFRJ).
Nos cortes corados pela hematoxilina e eosina, as fibras musculares apresentavam-se, em geral, preservadas, notando-se apenas uma certa irregularidade no tamanho. Já nas fibras coradas pelo Tricrômico de Gomori, observou-se leve grau de atrofia em algumas fibras e fibras com aumento de granulação avermelhada, que foi melhor observado na reação para succinodesidrogenase, na qual tais fibras tiveram aumento da atividade oxidativa, sobretudo periférica.
Notou-se, também, acúmulo de gordura nestas fibras e não se observou aumento de glicogênio nos cortes corados pelo ácido periódico-Schiff (PAS). Nas reações para ATPase, o padrão de distribuição das fibras em mosaico está presente. Estas alterações observadas foram descritas como “mitocondriopatia compatível com Síndrome de Kearns-Sayre”. Iniciou-se, então, o uso de coenzima Q10.
Após um ano, o paciente evoluiu com paralisia completa da musculatura extraocular, limitação funcional importante, e aos 19 anos de idade foi realizada correção cirúrgica da ptose palpebral (Figuras 1 e 2). Após a cirurgia, o diâmetro ocular da fenda palpebral passou de 5 mm em olho direito para 9 mm e de 4 mm para 8 mm em olho esquerdo.


Apresentou, à mesma época, alteração do timbre de voz (disfonia), quadro de fadiga, dispneia aos grandes esforços e hipoacusia. Além disso, apresentava episódios recorrentes de dispneia aos médios esforços, síncope, fadiga intensa e cefaleia. Em 2008, recebeu o diagnóstico de glaucoma primário de ângulo aberto, e foi revelada retinopatia com padrão de dispersão pigmentar associada à síndrome de Kearns-Sayre.
Aos 28 anos, após crise de dispneia aos médios esforços e episódio de síncope, foi solicitado o acompanhamento cardíaco, que revelou presença de distúrbio de condução elétrica cardíaca e dissociação atrioventricular total (BAVT) com frequência cardíaca entre 33-43 bpm, o que justificou a implantação de marca-passo permanente (Figuras 3 e 4).
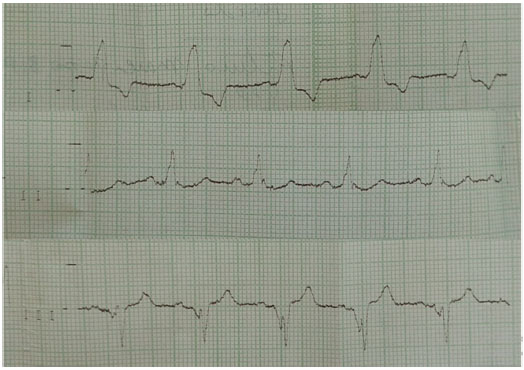

Ao exame clínico, encontrava-se orientado no tempo e no espaço, cooperativo, hidratado, normocorado, acianótico, sinais vitais estáveis com frequência cardíaca de 80 bpm, frequência respiratória de 16 irpm, pressão arterial sistêmica de 110/70 mmHg. Exame cardiovascular com Ictus palpável em duas polpas digitais, localizado na linha hemiclavicular à esquerda, no 5º espaço intercostal; ausência de pulsação visível no pericárdio; ausculta cardíaca com ritmo cardíaco regular em 2 tempos, bulhas normofonéticas, presença de sopro sistólico em foco mitral (++/6+).
Aparelho respiratório com ausência de deformidades e ausência de sinais de esforço respiratórios, frêmito toracovocal preservado, murmúrio vesicular bilateralmente audível, com ausência de ruídos adventícios e sem prejuízo da expansibilidade. Abdome flácido, peristáltico, indolor à palpação superficial e profunda, sem massas palpáveis ou visceromegalias. Membros superiores e inferiores com pulsos palpáveis simetricamente, com ausência de edemas e lesões visíveis, e reflexos preservados.
Na revisão de sistemas, foi detectada a presença da ptose palpebral bilateral, disfonia (voz hipernasal) e perfil auditivo compatível com perda sensorioneural leve/moderada à esquerda e leve/profunda à direita, confirmada através de impedanciometria.
Ao exame oftalmológico, apresentou acuidade visual OD= 20/25 e OE=20/60. Biomicroscopia de segmento anterior em OD e OE com conjuntivas claras, córnea transparente com ceratopatia superficial punctata leve inferior, câmara anterior ampla, sem reação inflamatória, pupila regular e normorreagente, e cristalino transparente.
Tonometria AO= 14 mmHg. Importante limitação da mobilidade muscular extrínseca no teste de versões e ducções e ausência do reflexo de Bell. À Fundoscopia, OD e OE apresentaram intensa dispersão do epitélio pigmentar da retina e áreas de atrofia de retina, disco óptico com bordos livres e escavação 0,9 x 0,9, concêntrica e com presença de estrias de Susanna em OD e 0,8 x 0,8 e concêntrica em OE; arteríolas de calibres reduzidos e trajetos normais. Faz uso de VIDISIC® gel e FRESH TEARS® colírio.
DISCUSSÃO
O diagnóstico da síndrome é clínico. As suspeitas começam com os sinais prévios da doença, como o surgimento de ptose palpebral, oftalmoplegia e retinopatia pigmentar antes da segunda década de vida. Cerca de 57% dos pacientes com a síndrome apresentam sinais de disfunção cardíaca e, assim, o acompanhamento cardíaco é fundamental devido ao risco associado de bloqueio de condução atrioventricular e morte súbita.
A SKS tem como fator causal a disfunção mitocondrial, consequente a mutações espontâneas. Têm-se, assim, alterações no DNA mitocondrial (mtDNA). Embora seja conhecido que o mtDNA é transmitido da mãe à descendência, os mecanismos estão sendo esclarecidos há pouco tempo. Sabe-se, nesse sentido, que em contraste com a abundância de mitocôndrias presentes em cada oócito, os espermatozoides contêm poucas mitocôndrias, e estas não são transmitidas à prole. Tem-se, portanto, que o mtDNA é de herança materna, e não paterna.4
A maioria das mutações são deleções no mtDNA que ocorrem de forma esporádica em períodos anteriores à embriogênese, como mutações em células germinativas, bem como durante o desenvolvimento inicial da vida, no período embrionário.4 O tamanho da deleção é variável, podendo ser 1,3kb a 8kb, ocorrendo também em posições variáveis no genoma mitocondrial. ⅔ das deleções acontecem entre as posições 8469 e 13147 do genoma, sendo o tamanho mais comumente observado de 4,9kb. Embora o tamanho das deleções varie, elas produzem fenótipos semelhantes.4
A necessidade da biópsia é controversa,5 apesar de ser necessária para confirmação diagnóstica, visto que possibilita a visualização de achados significativos nas fibras musculares. Outras alterações perceptíveis são a presença de manchas de Gömöri entre as fibras (visualizado por microscopia óptica), bem como a presença demasiada de mitocôndrias. Tem-se, aqui, uma relação direta: quanto maior o número de mitocôndrias na região da biópsia, maior será a probabilidade de encontrar uma mutação.6
É possível visualizar, também, o sinal de fibras vermelho áspero (fibras do tipo “ragged red”), que consiste na presença de uma coloração vermelho escuro por entre as fibras musculares, revelando uma doença mitocondrial. Outras análises podem ser feitas no fragmento, como: análises imuno-histoquímicas, que são mais específicas para o conteúdo enzimático mitocondrial; análise histoquímica do tecido muscular (como atividade enzimática da cadeia de transporte de elétron); e análise do DNA mitocondrial.6
Por sua característica multissistêmica,3,7 a análise e a correlação dos sinais e sintomas da síndrome devem ser feitas de forma minuciosa. No sistema cardiovascular, por exemplo, há um importante número de alterações que podem ser correlacionadas, como visto no estudo retrospectivo feito por Khambatta et al., realizado com 35 pacientes com SKS que tinham característica de apresentações cardiovasculares.8 Seis pacientes apresentavam síncope (17%); quatro pacientes tiveram morte repentina de causa cardíaca (11%). Investigações posteriores sugeriram alterações diversas cardíacas que poderiam indicar a utilização de um marca-passo como tratamento profilático, como bloqueio de ramo (11 pacientes apresentavam a alteração) e atraso de condução (23 pacientes; 66%). O paciente no presente relato apresentava bloqueio de ramo atrioventricular, como 31% dos pacientes do estudo de Khambatta et al., justificando assim a utilização de marca-passos.
A SKS não tem tratamento definitivo, havendo até o momento apenas o paliativo. A coenzima Q10(CoQ10) é o pilar nesta terapêutica. A CoQ10 tem sido intensamente estudada desde sua descoberta, em 1957. É um componente da cadeia de transporte eletrônico e participa da respiração aeróbica celular, gerando energia na forma de trifosfato de adenosina (ATP). A propriedade da CoQ10 de atuar como antioxidante ou pró-oxidante sugere papel importante na modulação do estado redox celular sob condições fisiológicas e patológicas, desempenhando, também, papel no processo de envelhecimento. Em vários modelos animais de doenças neurodegenerativas, a CoQ10 mostrou efeitos benéficos na redução do curso da doença, embora os efeitos sejam efêmeros.7
Entretanto, existem divergências sobre a eficácia da CoQ10. Um estudo randomizado, prospectivo, duplo-cego com 15 pacientes portadores de SKS e oftalmoplegia externa crônica progressiva utilizou a CoQ10 versus placebo e não apresentou resultados satisfatórios na disfunção muscular em um pequeno grupo de pacientes com a síndrome.8
O bloqueio AV frequentemente é precedido por distúrbios da condução intraventricular, como bloqueio de ramo esquerdo ou BRD isolado. A presença de BAVT é importante fator prognóstico nesta síndrome. Em revisão feita por Berenberg et al., apud Marie et al., incluindo cinco casos de sua casuística e mais 30 da literatura, BAVT foi a causa de morte em 20% dos pacientes.9
A grande maioria dos estudos mostrou melhora clínica, e estes são mais recentes quando comparados com estudos que não demonstraram a eficácia da CoQ10. No caso relatado, ocorreu melhora clínica significativa dos sintomas de fadiga muscular relatados pelo paciente e contribuiu para uma sutil melhora do quadro de oftalmoplegia. Entretanto, são necessárias mais evidências a fim de aprimorar o tratamento e proporcionar uma melhor qualidade de vida aos pacientes portadores da SKS.
CONCLUSÃO
A SKS é rara e, portanto, o médico deve possuir grande suspeição clínica para o diagnóstico da doença. Para isso, faz-se necessário o conhecimento das suas principais manifestações, bem como os sinais e sintomas associados, a fim de que haja o diagnóstico precoce, reduzindo, assim, os riscos inerentes à sua evolução, sendo o comprometimento cardiovascular com BAVT o responsável pela elevada mortalidade da doença.
Além disso, fazem-se necessários novos estudos a fim de aprimorar o seu tratamento e prevenir suas complicações. É importante perceber que a SKS é uma desordem complexa e, portanto, demanda o envolvimento de várias especialidades médicas, como neurologia, cardiologia, oftalmologia e endocrinologia. É de suma importância, pois, que os pacientes tenham um acompanhamento regular nestas áreas, haja vista que o prognóstico é reservado e a desordem é geralmente progressiva.
REFERÊNCIAS
1. De Castro Junior H, Pena FM, Ribeiro ML, Martins WA. Síndrome de Kearns-Sayre: Relato de caso. Insuf Card. 2011;6(2):92-5.
2. Nasseh IE, Tengan CH, Kiyomoto BH, Gabbai AA. Doenças mitocondriais. Rev Neurociênc. 2001;9(2):60-9.
3. von Blotzheim SG, Borruat, FX; Hirt L. Myopathies mitochondriales Oculaires: un éventail de présentations cliniques. Klin Monatsbl Augenheilkd. 1998;212(5):299-300.
4. Ribeiro EM, Vasconcelos DO. Mitochondrial diseases. Rev Bras Med. 2006;63(4):169-73.
5. Zierz S, Jahns G, Jerusalem F. Coenzyme Q in serum and muscle of 5 patients with Kearns-Sayre syndrome and 12 patients with ophthalmoplegia plus. J Neurol. 1989;236(2):97-101. DOI: 10.1007/BF00314404
6. Rubin RM, Sadun AA. Ocular Myopathies. In: Yanoff M, Duker J. Ophthalmology. 3rd ed. Saint Louis: Mosby; 2008.
7. Santos GC, Antunes LMG, Santos AC, Bianchi MLP. Coenzyme Q10 and its effects in the treatment of neurodegenerative diseases. Braz J Pharm Sci. 2009;45(4):607-18. DOI: 10.1590/S1984-82502009000400002
8. Khambatta S, Nguyen DL, Beckman TJ, Wittich CM. Kearns-Sayre syndrome: a casa series of 35 adults and children. Int J Gen Med. 2014;7:325-32.
9. Marie SKN, Carvalho AAS, Fonseca LF, Carvalho MS, Reed UC, Scaff M. Kearns-Sayre syndrome “plus”: classical clinical findings and dystonia. Arq Neuro-Psiquiatr. 1999;57(4):1017-23. DOI: 10.1590/S0004-282X1999000600020





Fonte de financiamento: declaram não haver.
Parecer CEP: não aplicável.
Conflito de interesses: declaram não haver.
Recebido em:
8 de Agosto de 2017.
Aceito em:
6 de Dezembro de 2017.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados
 Ler em inglês
Ler em inglês
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket