

João Vitor de Oliveira Pereira1,2,3; Ana Luiza Machado Ribeiro Pimentel1,2,3; Dillan Cunha Amaral2,4; Ricardo Noguera Louzada4,5; Luís Alexandre Rassi Gabriel3
DOI: 10.17545/eOftalmo/2024.0009
RESUMO
O estudo relata um caso ultrarraro de atrofia girata de coroide e retina com variante de significado incerto homozigota no gene OAT (c.416T>G:p.M139R). O caso é de uma paciente de 59 anos com sintomas desde a infância, apresentando acuidade visual de percepção luminosa em olho direito e de 20/100 em olho esquerdo. Ao fundo de olho observou-se presença de atrofia coriorretiniana característica da atrofia girata com atenuação moderada dos calibres arteriovenulares. Foram realizados exames adicionais como biomicroscopia oftálmica de segmento anterior, retinografia/autofluorescência, perimetria visual cinética semiautomática de Goldmann e adaptometria ao escuro. Nos exames adicionais, foram registradas alterações anatômicas e disfuncionais como múltiplas áreas de atrofia girata coriorretinianas, espículas ósseas, atenuação moderada dos calibres arteriolovenulares e atrofia peridiscal no olho esquerdo. A paciente foi encaminhada para acompanhamento nutricional para tratamento com dieta restritiva de arginina e suplementação com vitamina B6.
Palavras-chave: Atrofia girata; OAT; Vitamina B6; Biomarcador; Análise de DNA; Genética molecular; Genética oftálmica; Ornitina.
ABSTRACT
This report presents an exceptionally rare case of gyrate atrophy affecting the choroid and retina, accompanied by a homozygous variant of uncertain significance in the OAT gene (c.416T>G:p.M139R), which is the first documented instance in ClinVar. The case involves a 59-year-old patient who has experienced symptoms since childhood. Visual acuity was reported as light perception in the right eye and 20/100 in the left eye. Fundoscopic examination revealed characteristic chorioretinal atrophy of gyrate atrophy, along with moderate attenuation of arteriolovenular calibers. Additional diagnostic tests included ophthalmic biomicroscopy of the anterior segment, retinography/autofluorescence, Goldmann semiautomated kinetic perimetry, and dark adaptometry. Further examinations unveiled anatomical and functional abnormalities in the left eye, including multiple areas of chorioretinal gyrate atrophy, bone spicules, moderate arteriolovenular caliber attenuation, and peridiscal atrophy. The patient was subsequently referred for nutritional monitoring and prescribed a restrictive arginine diet along with vitamin B6 supplementation as part of the treatment plan.
Keywords: Gyrate atrophy; OAT; Vitamin B6; Biomarker; DNA analysis; Molecular genetics; Ophthalmic genetics; Ornithine.
INTRODUÇÃO
A atrofia girata de coroide e retina (AGCR) consiste em uma rara doença genética hereditária autossômica recessiva1. A etiologia advém de mutação bialélica no gene OAT, levando à deficiência da ornitina aminotransferase que, por sua vez, ocasiona hiperornitinemia2,3. A enzima ornitina aminotransferase (OAT) é uma enzima mitocondrial que converte a ornitina (derivada da arginina) em prolina. A deficiência dessa enzima pode levar a um acúmulo de ornitina tanto no plasma quanto nos tecidos. O acúmulo de ornitina na retina pode exercer um efeito citotóxico no epitélio pigmentar da retina (EPR)4. Os sintomas podem incluir nictalopia, constrição perimétrica e diminuição de acuidade visual. Os sinais oculares incluem múltiplas áreas confluentes de atrofia de coroide e retina, de cor creme-amarelada confluentes centripetamente e com bordos circulares bem definidos, por isto o termo girata, oriundo do vocábulo grego gyros, que significa círculo5,6. Adicionalmente, notam-se no fundo de olho as espículas ósseas, atenuação dos calibres de arteríolas e vênulas, além de atrofia peridiscal. São comuns também catarata precoce e miopia7. As alterações sensoriais e motoras discretas podem também ocorrer na avaliação extraocular. A hiperornitinemia pode ser encontrada em pacientes diagnosticados com atrofia girata, uma vez que, a mutação no gene OAT bloqueia a conversão desse aminoácido2,8. A idade de início da doença é variável e a fisiopatologia não é bem conhecida, mas sabe-se que a doença tem início com a degeneração do epitélio pigmentar da retina (EPR).
Nesse estudo relatamos o primeiro caso de paciente com diagnóstico clínico de atrofia girata associada a variante de significado incerto c.416T>G no gene OAT.
RELATO DE CASO
Paciente do sexo feminino de 59 anos de idade, chegou à clínica para acompanhamento de AGCR, sintomática desde a infância apresentando acuidade visual em olho direito (OD) de percepção luminosa e, em olho esquerdo (OE), de 20/100. Ao exame de biomicroscopia, observado a pseudofácica bilateralmente. Realizados reflexos pupilares e de motilidade ocular que apresentaram normais em ambos os olhos.
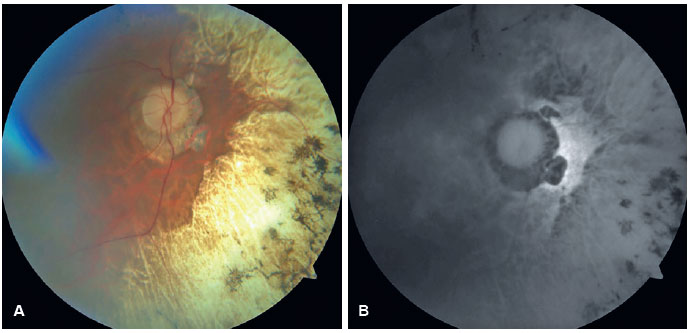
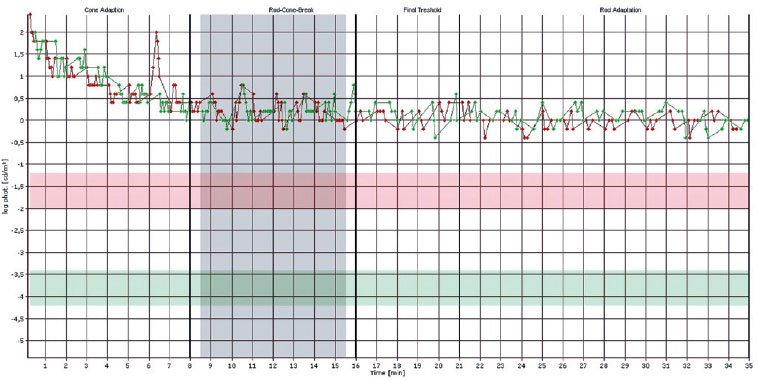
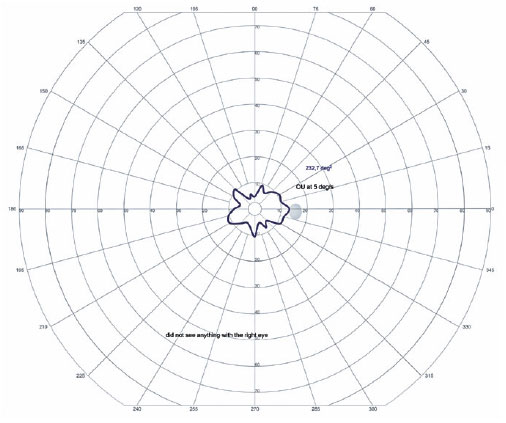
Em OD, possui nistagmo pendular proveniente de sua baixa visão. Ao exame de fundo de olho de ambos os olhos apresentou múltiplas áreas de atrofia coriorretiniana, espículas ósseas, atenuação moderada dos calibres arteriolovenulares e atrofia peridiscal (Figura 1). Na autofluorescência de OE, tem hipoautofluorescência nas áreas de atrofia. Em seu exame clínico verificou-se nictalopia acentuada, mensurada em adaptometria ao escuro, necessitando de intensidade luminosa aproximadamente 10.000 vezes maior que um indivíduo com integridade funcional de sua retina (Figura 2), constrição perimétrica, aferida em perimetria visual cinética semiautomática de Goldmann (PVCSG) (Figura 3).
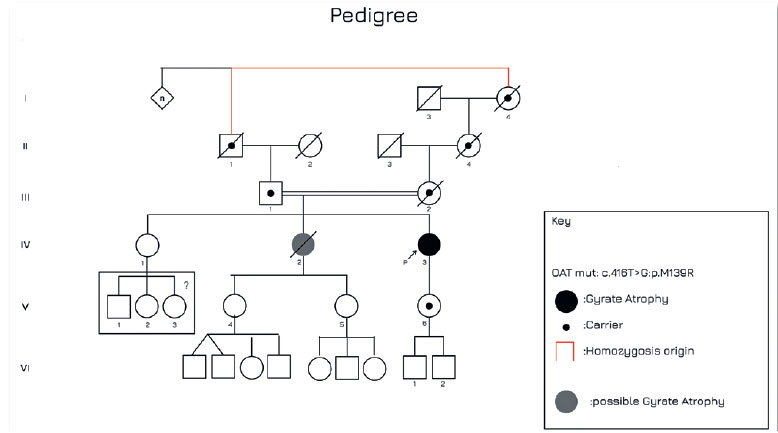
O teste genético identificou uma variante homozigota classificada como sendo de significado incerto c.416T>G:p.M139R (Clinvar:VCV001027070.8) no gene OAT. Foi realizada segregação do pai e foi possível detectar a mesma variante em heterozigose, conforme pode ser visto no heredograma (Figura 4). Os softwares de bioinformática SIFT, PolyPhen-2, Align-GVGD todos indicam que esta variante genética é teoricamente deletéria e o banco de dados genético gnomAD mostra frequência desta variante igual a 0.003% em população de mais de 125.000 indivíduos sequenciados. A paciente foi encaminhada para avaliação de suplementação de vitamina B6 e amino ácidos essenciais além da prescrição de uma dieta restritiva de arginina.
Foi realizado um sequenciamento de nova geração através de um painel comercial com 328 genes com certificação internacional. O sequenciamento Sanger foi realizado para confirmar as variantes encontradas. A análise da segregação paterna da paciente foi realizada. No exame da PVCSG, utilizou-se o aparelho Octopus 900 com software Eye Suite versão i9.6.3.0 - Haag-Streit Diagnostics, com projeção luminosa circular de diâmetro de 9 mm (tamanho V), intensidade luminosa de 318 cd/m2 (4e), à velocidade constante de 5º/s, vetores centrípetros em cúpula Ganzfeld com 31,4 asb de luminância.
Outros testes como biomicroscopia oftálmica de segmento anterior, retinografia/ autofluorescência e adaptometria ao escuro foram realizados pelo mesmo oftalmologista usando os respectivos aparelhos: biomicroscópio BQ 900 - Haag-Streit International, Cirrus Photo 800 – Zeiss e DARKadaptometer – Roland Consult.
DISCUSSÃO
Este estudo relata características clínicas e genéticas de uma paciente com AGCR com uma variante de significado incerto c.416T>G:p.M139R de forma bialélica. A paciente possuí o diagnóstico clínico é de atrofia girata, entretanto o teste molecular não é totalmente conclusivo.
Atualmente, as correlações genótipo-fenótipo em AGCR ainda não estão claramente definidas1. Em outro estudo, Katagiri et al. avaliou que a mutação no gene OAT está associada a dois fenótipos clínicos diferentes. A maioria dos pacientes apresenta atrofia girata, na qual se tem elevados de ornitina plasmática e degeneração coriorretiniana. Já no outro fenótipo conhecido para a mutação, alguns pacientes apresentam um distúrbio mais grave, caracterizado por início neonatal de hiperamonemia, vômitos e sinais de encefalopatia. Esses pacientes geralmente apresentam baixos níveis de ornitina no plasma, indicando que no período neonatal a reação catalisada pela OAT é a formação de ornitina2.
Atualmente, três causas de hiperornitinemia são reconhecidas: a síndrome de hiperornitinemia-hiperamonemia-homocitrullinuria, a deficiência de Δ(1)-Pirrolina-5-carboxilato sintetase e a atrofia girata. A paciente não exibe sintomas compatíveis com as duas primeiras condições mencionadas, o que reforça o diagnóstico de atrofia girata com base na apresentação clínica e nos resultados do teste genético.
O diagnóstico anatômico e sindrômico da AGCR no caso em questão é típico, entretanto, o diagnóstico etiológico traz uma variante de significado incerto, que levanta a questão de talvez se tratar de uma nova variante. Apesar de existir um código de identificação para esta variante no website Clinvar (https://www.ncbi.nlm.nih.gov/clinvar/variation/1027070/), em que a primeira identificação notada ocorreu em 2020, porém com um diagnóstico de retinose pigmentar, não há dados clínicos publicados acerca do caso mencionado.
O mecanismo pelo qual a deficiência de ornitina d-aminotransferase e o conseqüente acúmulo de ornitina levam à degeneração coriorretiniana não é totalmente compreendido. O acúmulo de ornitina ou seus produtos metabólicos podem causar danos diretos aos tecidos e o EPR é o local inicial do dano em pacientes com atrofia girata5. Além disso, estudos em modelos animais mostraram que a redução crônica dos níveis plasmáticos de ornitina pode prevenir a degeneração retiniana e o acúmulo de ornitina pode levar a degeneração do EPR7-9.
O tratamento clássico para a AGCR é a prescrição de vitamina B6 ou piridoxina com aminoácidos essenciais e uma dieta restritiva de arginina, visto que a arginina é a precurssora da ornitina10. Um estudo prévio observou que a implementação precoce de um tratamento dietético restritivo de arginina tem o efeito aparente de retardar a progressão das lesões coriorretinianas. Além disso, notaram que esse tratamento resultou em uma aparência única do fundo do olho, assemelhando-se a um estágio inicial de retinose pigmentar3. No entanto, a eficácia desse método é debatida, com alguns estudos relatando melhorias ou estabilização das mudanças na retina, enquanto outros indicam a progressão das complicações coriorretinianas. A adoção desse método requer um acompanhamento minuscioso. Diferenças na idade de início do tratamento, na adesão à dieta e a variabilidade genética na AGCR provavelmente desempenham um papel importante nessa diversidade de resultados10-14.
Nesse caso, a paciente realizou a dieta com restrição do aminoácido ornitina combinada com a suplementação da vitamina B6, uma precursora do piridoxal fosfato, que é uma coenzima da ornitina aminotransferase15.
A lenta progressão das alterações degenerativas coriorretinianas, típicas da AGCR, torna muito difícil a avaliação da eficácia de qualquer abordagem terapêutica para esta doença16.
Famílias consanguíneas representam um fator de risco para aumento da incidência de doenças genéticas e para tal é de extrema importância que seja realizado o aconselhamento genético diante do planejamento familiar de casais consanguíneos17.
Em conclusão, nesse estudo, foi relatado o primeiro caso de paciente com diagnóstico clínico de atrofia girata associado a variante homozigota c.416T>G (p.M139R) classificada como sendo de significado incerto (ClinVar variation ID: 1027070) no gene OAT.
REFERÊNCIAS
1. Doimo M, Desbats MA, Baldoin MC, Lenzini E, Basso G, Murphy E, et al. Functional analysis of missense mutations of OAT, causing gyrate atrophy of choroid and retina. Hum Mutat. 2013;34(1):229-36.
2. Katagiri S, Gekka T, Hayashi T, Ida H, Ohashi T, Eto Y, et al. OAT mutations and clinical features in two Japanese brothers with gyrate atrophy of the choroid and retina. Doc Ophthalmol. 2014;128(2):137-48.
3. Kaiser-Kupfer MI, Caruso RC, Valle D. Gyrate atrophy of the choroid and retina: further experience with long-term reduction of ornithine levels in children. Arch Ophthalmol. 2002;120(2):146-53.
4. Valle D, Kaiser-Kupfer MI, Del Valle LA. Gyrate atrophy of the choroid and retina: deficiency of ornithine aminotransferase in transformed lymphocytes. Proc Natl Acad Sci U S A. 1977;74(11):5159-61.
5. Takki KK, Milton RC. The natural history of gyrate atrophy of the choroid and retina. Ophthalmology. 1981;88(4):292-301.
6. Berson EL, Schmidt SY, Shih VE. Ocular and biochemical abnormalities in gyrate atrophy of the choroid and retina. Ophthalmology. 1978;85(10):1018-27.
7. Kuwabara T, Ishikawa Y, Kaiser-Kupfer MI. Experimental model of gyrate atrophy in animals. Ophthalmology. 1981;88(4):331-5.
8. Wang T, Steel G, Milam AH, Valle D. Correction of ornithine accumulation prevents retinal degeneration in a mouse model of gyrate atrophy of the choroid and retina. Proc Natl Acad Sci U S A. 2000;97(3):1224-9.
9. Wang T, Milam AH, Steel G, Valle D. A mouse model of gyrate atrophy of the choroid and retina. Early retinal pigment epithelium damage and progressive retinal degeneration. J Clin invest. 1996;97(12):2753-62.
10. Balfoort BM, Buijs MJN, Asbroek ALMAT, Bergen AAB, Boon CJF, Ferreira EA, et al. A review of treatment modalities in gyrate atrophy of the choroid and retina (GACR). Mol Genet Metab. 2021;134(1-2):96-116.
11. Vannas-Sulonen K, Simell O, Sipilä I. Gyrate atrophy of the choroid and retina. The ocular disease progresses in juvenile patients despite normal or near normal plasma ornithine concentration. Ophthalmology. 1987;94(11):1428-33.
12. Kaiser-Kupfer MI, Caruso RC, Valle D. Gyrate atrophy of the choroid and retina: long-term reduction of ornithine slows retinal degeneration. Arch Ophthalmol. 1991;109(11):1539-48.
13. Mashima YG, Weleber RG, Kennaway NG, Inana G. Genotype-phenotype correlation of a pyridoxine-responsive form of gyrate atrophy. Ophthalmic Genet. 1999;20(4):219-24.
14. Ramesh V, McClatchey AI, Ramesh N, Benoit LA, Berson EL, Shih VE, et al. Molecular basis of ornithine aminotransferase deficiency in B-6-responsive and-nonresponsive forms of gyrate atrophy. Proc Natl Acad Sci U S A. 1988;85(11):3777-80.
15. Ohkubo Y, Ueta A, Ito T, Sumi S, Yamada M, Ozawa K, et al. Vitamin B6-responsive ornithine aminotransferase deficiency with a novel mutation G237D. Tohoku J Exp Med. 2005;205(4):335-42.
16. Santinelli R, Costagliola C, Tolone C, D'Aloia A, D'Avanzo A, Prisco F, et al. Low-protein diet and progression of retinal degeneration in gyrate atrophy of the choroid and retina: A twenty-six-year follow-up. J Inherit Metab Dis. 2004;27(2):187-96.
17. Huang J, Fu J, Fu S, Yang L, Nie K, Duan C, et al. Diagnostic value of a combination of next-generation sequencing, chorioretinal imaging and metabolic analysis: lessons from a consanguineous Chinese family with gyrate atrophy of the choroid and retina stemming from a novel OAT variant. Br J Ophthalmol. 2019; 103(3):428-35.
INFORMAÇÃO DOS AUTORES |
|
 |
» João Vitor de Oliveira Pereira https://orcid.org/0000-0001-8806-0689 http://lattes.cnpq.br/5419937219657840 |
 |
» Ana Luiza Machado Ribeiro Pimentel https://orcid.org/0000-0003-1122-6657 http://lattes.cnpq.br/8010513090617384 |
 |
» Dillan Cunha Amaral https://orcid.org/0009-0002-7948-154X https://lattes.cnpq.br/7959357721386149 |
 |
» Ricardo Noguera Louzada https://orcid.org/0000000296105768 http://lattes.cnpq.br/5978866539118374 |
 |
» Luís Alexandre Gabriel Rassi https://orcid.org/0000000204587131 http://lattes.cnpq.br/3226810848914264 |
Financiamento: Declaram não haver.
Conflitos de Interesse: Declaram não haver.
Recebido em:
27 de Fevereiro de 2023.
Aceito em:
21 de Janeiro de 2024.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados
 Ler em inglês
Ler em inglês
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket