

Felipe Di Domenico1; Felipe Kenzo Ishida2; Murilo Simão Cenovicz3
DOI: 10.17545/e-oftalmo.cbo/2016.73
RESUMO
A doença de Stargardt é uma rara distrofia macular de início precoce que afeta progressivamente a acuidade visual central. Na maior parte das vezes é causada por uma herança autossômica recessiva. Apresentamos o caso de uma família com doença de Stargardt causada por herança autossômica dominante, descrevendo sua história, achados clínicos e exames complementares relevantes.
Palavras-chave: Degeneração Macular; Células Fotorreceptoras Retinianas Cones; Distrofia Macular; Proteína humana ELOVL4
ABSTRACT
Stargardt disease is a rare early-onset macular dystrophy, which progressively affects central visual acuity. In most cases, it is caused by an autosomal recessive inheritance. Here, we present the case of a family with Stargardt disease caused by autosomal dominant inheritance, together with a description of their history, clinical findings, and pertinent complementary examinations.
Keywords: Macular Degeneration: Retinal Cone Photoreceptor Cells: Macular Dystrophy: ELOVL4 protein, human
INTRODUÇÃO
Doença de Stargardt (STGD) é a distrofia macular recessiva mais comum e é caracterizada pela diminuição da visão central, atrofia da mácula e do epitélio pigmentar da retina subjacente e presença frequente de manchas amareladas (flecks) no polo posterior da retina1.
As características clínicas da doença variam de acordo com a severidade do seu genótipo e com a sensibilidade dos cones foveais e do epitélio pigmentado da retina (EPR) a esse genótipo. A baixa de acuidade visual é a manifestação clínica mais comum podendo variar de 20/30 a 20/200, enquanto a idade de aparecimento pode variar de 5 a 50 anos de idade2.
Os achados de fundo de olho incluem manchas amareladas esparsas denominadas flecks (refletem o acúmulo de bisretinoide subjacente ao EPR) e atrofia do EPR na região macular, descrita como aspecto de “cobre batido” 2. Na angiofluoresceinografia (AF), o bisretinoide acumulado adjacente à coroide bloqueia a excitação luminosa do corante situado na circulação coroidea, achado denominado silêncio coroideo - deixando assim os vasos retinianos em evidência banhados em um fundo hipofluorescente 3,4. Observase também atrofia do EPR, maculopatia em “olho de boi”, “flecks” hiperfluorescentes e preservação do EPR peridiscal (“peripapillary sparing”)5.
Geneticamente, STGD é uma desordem heterogênea herdada geralmente como uma desordem autossômica recessiva, mas raramente se apresenta como um traço autossômico dominante com um começo mais atrasado de sintomas clínicos6. Um lócus recessivo foi mapeado no cromossoma 1p (STGD tipo 1)7. Esse gene codifica uma proteína de transmembrana específica da retina, ABCA4, que pertence à família de transportadores de membrana do tipo ATP Binding Cassette (ABC)1. Poucos casos de famílias com diagnóstico de doença de Stargardt foram descritos com transmissão de característica autossômica dominante, como o mapeamento de um lócus no cromossomo 13q (STGD tipo 2)8, do cromossomo 6q (STGD tipo 3)9 e do cromossomo 4p (STGD tipo 4) 1.
A Distrofia macular dominante Stargardt-like (doença de Stargardt tipo 3) foi primeiramente descrita por Stone et al. em 1994 - uma grande família com um fenótipo semelhante a Stargardt - mapeando o gene no cromossomo 6q9. Zhang et al. em 2001 identificaram, em cinco famílias afetadas por essa doença, uma deleção de 5 pares de bases nitrogenadas no gene ELOVL4 do cromossomo 6q10. Bernstein et al., também em 2011, identificaram uma mutação heterozigótica complexa no gene ELOVL4 em que duas deleções 1-bp separadas por 4 nucleotídeos resultaram num desvio de enquadramento e truncamento da proteína ELOVL4 - mostrando que o fenótipo induzido por essa mutação é muito variado entre os pacientes11.
O gene ELOVL4 (sigla para Elongation Of Very Long Chain Fatty Acids - Alongamento de Ácidos Graxos de Cadeia muito Longa) codifica uma enzima biossintética no retículo endoplasmático responsável pela síntese de ácidos graxos com mais de 26 átomos de carbono. Este gene é expresso no cérebro e na retina - local onde a expressão está restringida às células fotorreceptoras. As mutações associadas à STGD tipo 3 causam um tráfego deficiente da proteína mutante, o que resulta em acúmulo de lipofuscina e em morte das células fotorreceptoras2.
As manifestações clínicas são muito semelhantes às encontradas na STGD tipo 1. Os depósitos de lipofuscina na STGD tipo 3 tendem a ser um pouco maiores que na STGD tipo 1, e frequentemente assumem uma aparência de “borboleta” nas imediações das lesões maculares2. O ERG é usualmente normal, e a angiofluoresceinografia mostra uma menor prevalência do achado de silêncio coroideo2.
A seguir é apresentado o caso de uma família diagnosticada com Doença de Stargardt tipo 3, a história da doença, achados clínicos e exames complementares relevantes.
RELATO DE CASO
PACIENTE 1
Paciente feminina, 47 anos. Queixa principal: “Embaçamento visual”.
História mórbida atual: Paciente relata piora da acuidade visual progressiva durante a vida e mais acentuada nos últimos 3 anos. Refere sintoma em ambos os olhos, com maior intensidade no olho direito.
Relata suspeita médica de “distrofia na retina” em outro serviço médico há cerca de 10 anos, sem posterior investigação.
Nega doenças sistêmicas prévias. Nega cirurgias e internamentos prévios. Nega alergias.
Exame Físico:
O exame oftalmológico constatou acuidade visual 20/400 no olho direito (OD) e 20/200 no olho esquerdo (OE), ambos com a melhor correção óptica.
Biomicroscopia sem alterações. Tonometria de 15 mmHg em ambos os olhos (AO).
Na avaliação fundoscópica observou-se atrofia difusa do epitélio pigmentado da retina com presença de “flecks” esparsos esvaecidos em ambos os olhos (Figura 1).
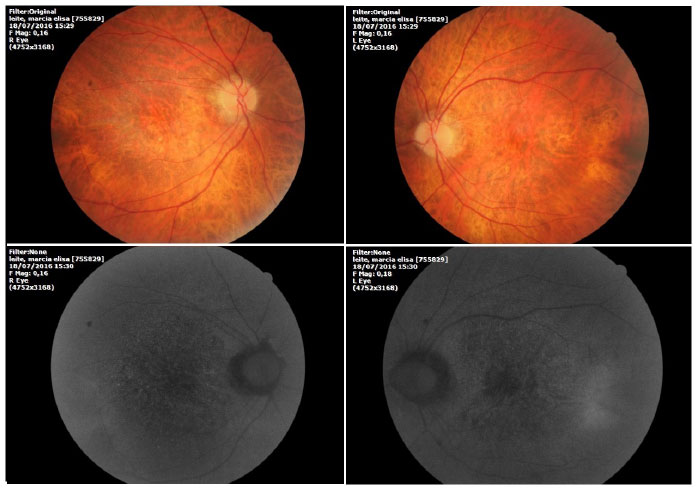
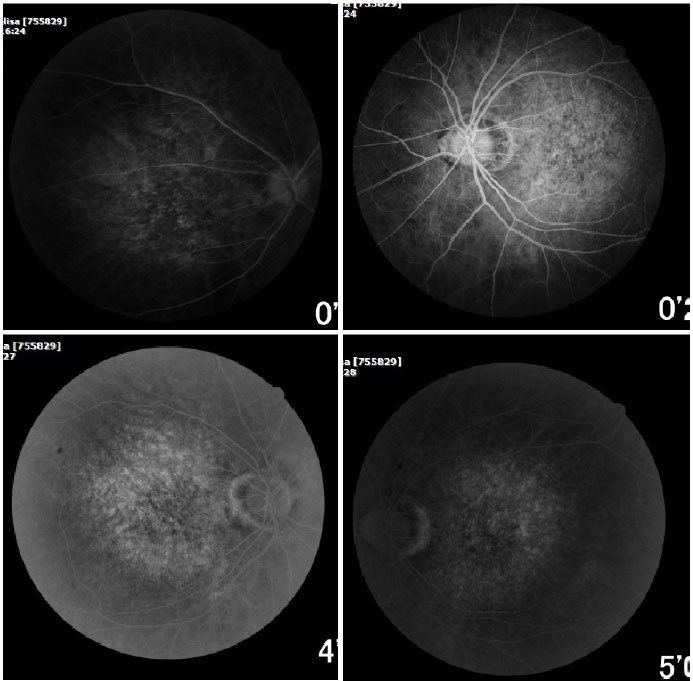
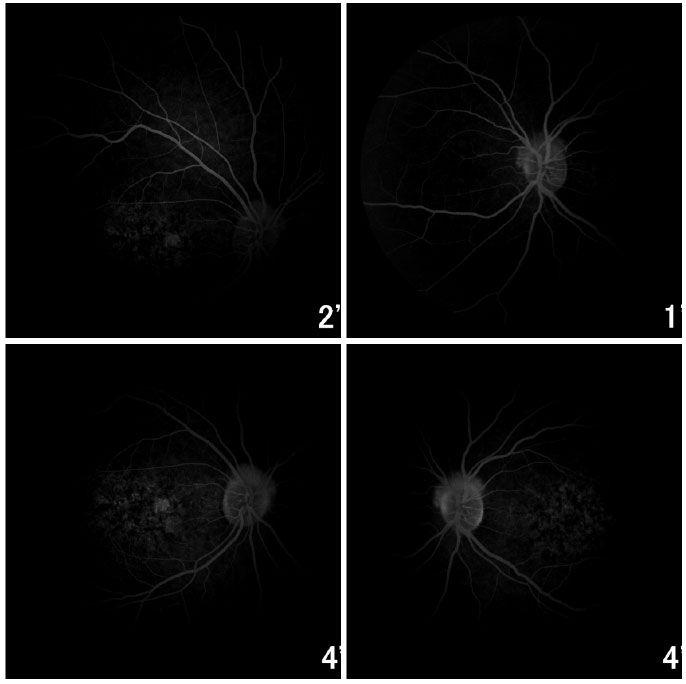
A angiofluoresceinografia demonstrou hiperfluorescência por defeito em janela na área macular e perimacular, com aspecto pontilhado coalescente de pontos hiperfluorescentes até fases tardias (Figura 2).
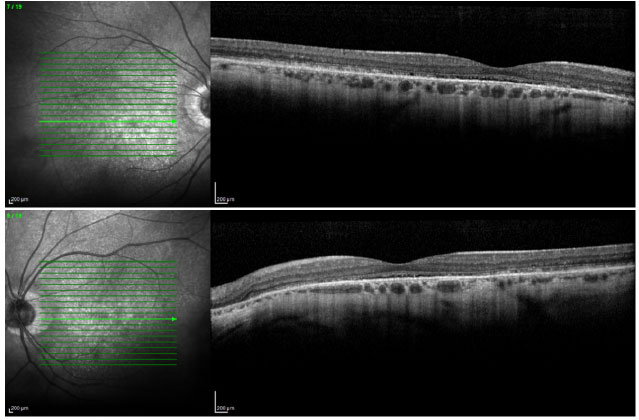
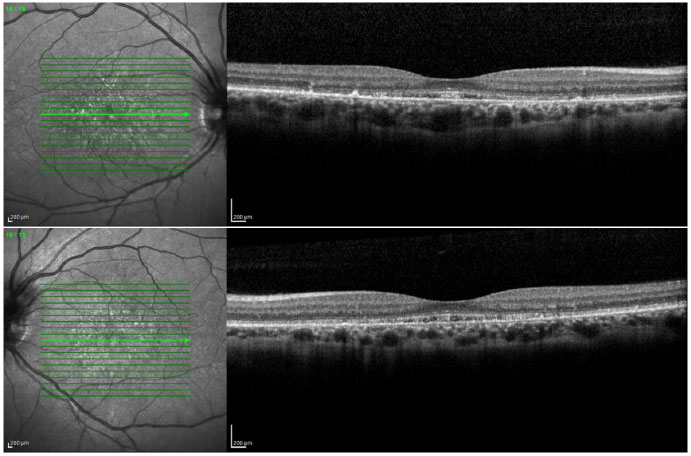
Ao exame da tomografia de coerência óptica foi visualizada alteração na camada elipsoide com perda de fotorreceptores (Figura 3).
INVESTIGAÇÃO FAMILIAR
PACIENTE 2
Irmão da paciente 1 vem ao serviço. Masculino, 39 anos. Traz exame de Eletrorretinograma com alteração fotópica. Ao questionarmos sobre sintomas oculares, paciente refere fotofobia intensa.
O exame oftalmológico constatou acuidade visual 20/30 no OD e 20/80 no OE, com a melhor correção óptica em AO. Biomicroscopia sem alterações. Tonometria de 12 mmHg em AO.
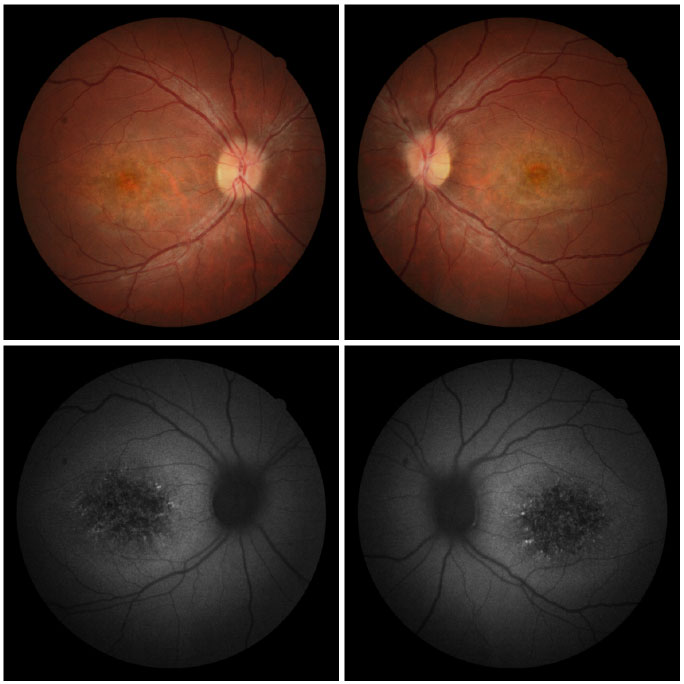
Na avaliação fundoscópica observou-se atrofia difusa do epitélio pigmentado da retina, mácula com aspecto de cobre batido e presença de flecks puntiformes na região perimacular em ambos os olhos (Figura 4 A-B).
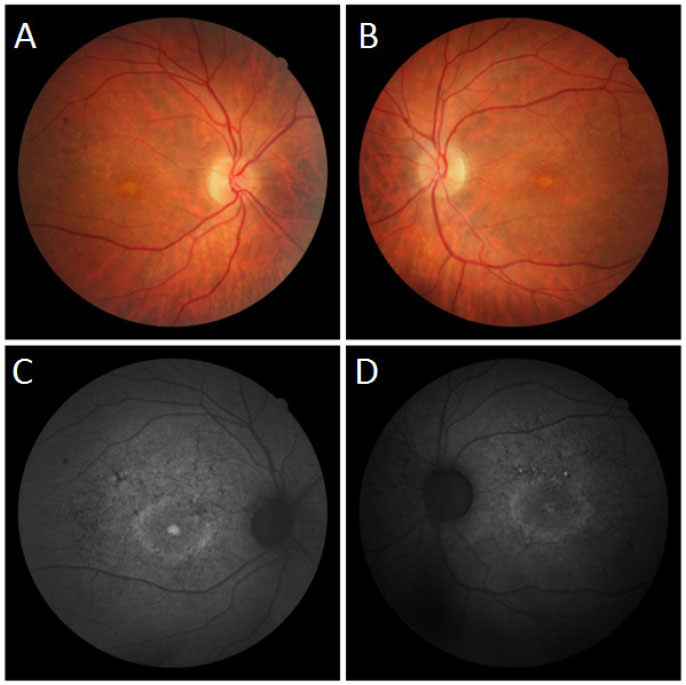
Nas imagens com filtro anerita observou-se maculopatia em bulls eye, dotada de áreas de hiperautofluorescência perimacular e foveal, assim como atrofias puntiformes do EPR central (Figura 4 C-D).
A angiofluoresceinografia demonstrou hiperfluorescência por defeito em janela na área macular e perimacular, com aspecto pontilhado coalescente de pontos hiperfluorescentes até fases tardias (Figura 5).
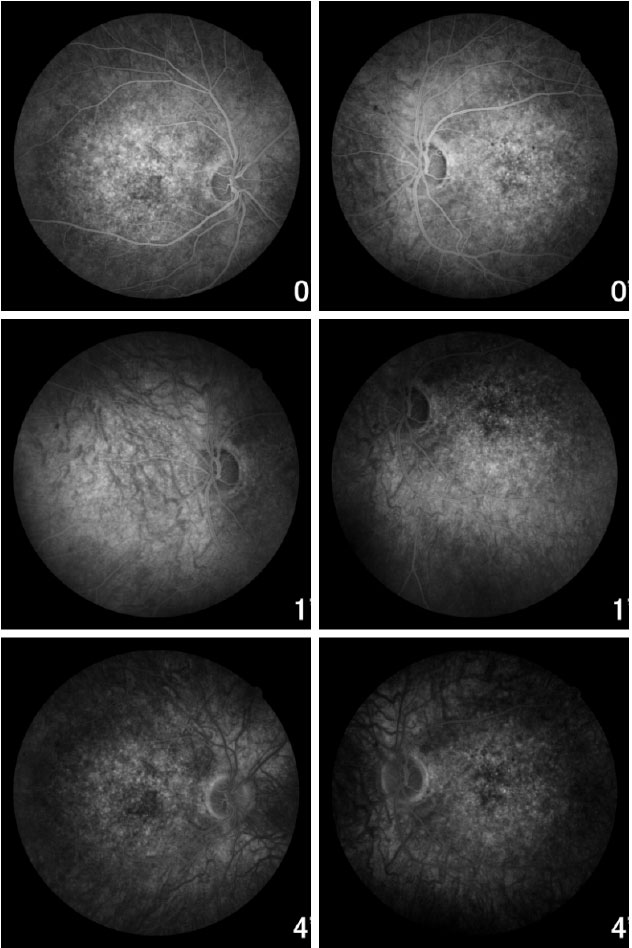
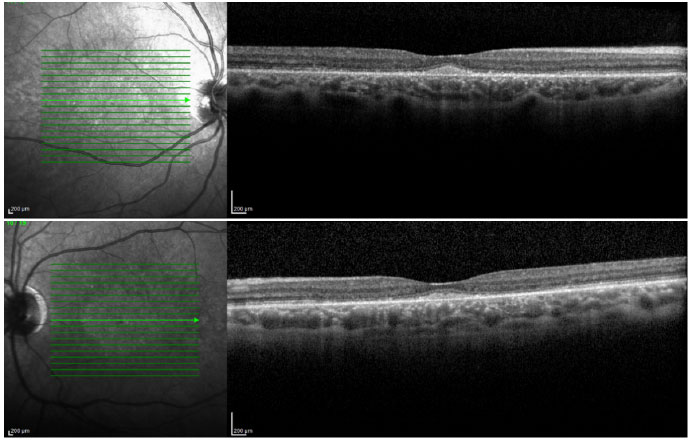
Ao exame da tomografia de coerência óptica observaram-se depósitos foveais e alteração da camada elipsoide dos fotorreceptores (Figura 6).
INVESTIGAÇÃO FAMILIAR
PACIENTE 3
Filha da paciente 1 vem ao serviço. Feminina, 29 anos. Nega sintomas oculares.
Relata possuir diagnóstico de Lúpus Eritematoso Sistêmico e Poliarterite Nodosa. Refere ter sofrido rotura traumática de ligamento do joelho há 3 semanas.
O exame oftalmológico constatou acuidade visual 20/20 no OD e 20/25 no OE com a melhor correção óptica em AO. Biomicroscopia sem alterações. Tonometria de 12 mmHg em AO.
Na avaliação fundoscópica observou-se aspecto de cobre batido na região macular, atrofia do epitélio pigmentado da retina na região da retina central e presença de “flecks” esparsos na região perimacular em AO (Figura 7).
A angiofluoresceinografia demonstrou intenso silêncio coroideo com contraste entre os vasos retinianos contra um fundo escuro da coroide. Observam-se áreas de hiperfluorescência por defeito em janela na área macular e perimacular em AO (Figura 8).
Ao exame da tomografia de coerência óptica observou-se presença de depósitos situados abaixo do EPR e alteração na camada elipsoide (Figura 9).
DISCUSSÃO
A doença de Stargardt de padrão recessivo é a distrofia macular hereditária mais comum, representando 7% das distrofias retinianas12. Já a distrofia macular Stargardt-like (STGD tipo 3) foi reportada em poucos relatos na literatura, sendo uma doença atípica até em serviços especializados.
O diagnóstico inicial da doença de Stargardt recessiva, segundo Fishman, é mais prevalente na faixa dos 20-29 anos13. Não foram achados dados em relação à idade de diagnóstico inicial em pacientes com doença de Stargardt autossômica dominante. Nossos pacientes tiveram o diagnóstico apenas no nosso serviço, ou seja, os 3 pacientes com 47, 39 e 29 anos respectivamente.
Pacientes com ST G apresentam com certa frequência o sintoma de fotofobia, e podem apresentam relato de melhor visão noturna14, sintoma relatado pelo paciente 2. Nossos pacientes tinham visão de cores normal, concordando com os dados da literatura. O sintoma de alteração de visão de cores normalmente não é encontrado nos pacientes com doença de Stargardt dominante14.
Edwards et al. encontraram acuidade visual em pacientes Stargardt tipo 3 variando de 20/20 a 20/800, com a maioria dos pacientes com idade acima de 30 anos tendo uma visão de 20/200 ou pior14. Após apresentar acuidade visual de 20/200, a progressão é lenta, diferente da doença de Stargardt recessiva. Tal dado entrou em conflito com os nossos achados; o paciente 2, de 39 anos, apresentou acuidade visual de 20/30 no OD e 20/80 no OE; a paciente 3, de 29 anos, apresentou acuidade visual de 20/20 no OD e 20/25 no OE. A paciente 1, de 47 anos, se enquadrou nos resultados de tal relato, com acuidade visual de 20/200 no OD e 20/400 no OE.
Edwards et al. estudaram 2 famílias com doença de Stargardt tipo 3, e encontraram padrões de aparição de alteração na fundoscopia conforme o estágio da doença14. Os achados iniciais incluíam atrofia subfoveal do epitélio pigmentado da retina associado a uma área de defeito em janela na angiofluoresceinografia. Com o passar dos anos haveria aumento da atrofia do EPR que se assemelha às lesões manchadas observadas em pacientes com doença de Stargardt recessiva. As manchas flecks estavam geralmente ausentes nessa fase. Ao longo de vários anos, as lesões evoluíram para uma área bem circunscrita de retina, atrofia epitélio pigmentar da retina e coroide de tamanho crescente associado com flecks na maioria dos pacientes. Uma aparência de metal batido da lesão atrófica estava frequentemente presente. Não tivemos a oportunidade de realizar um acompanhamento horizontal dos pacientes até o momento do estudo. No entanto, as alterações fundoscópicas dos três pacientes estão em concordância com os achados descritos por Edwards et al. 14
As famílias de distrofia macular de tipo Stargardt autossômica dominante ligada aos cromossomas 6q14 e 13q34 têm um fenótipo de fundo semelhante14. Segundo Edward et al., as famílias ligadas a 6q14 não têm a coroide escura observada na doença recessiva de Stargardt e não se sabe se as famílias ligadas ao 13q34 têm um silêncio coroideo. Lopez et al. descreveram uma família com distrofia macular de tipo Stargardt autossômica dominante que apresentaram silêncio coroideo15. Em nossos pacientes, observou-se silêncio coroideo na angiofluoresceinografia da paciente 3 (Figura 8). Ryan et al. relataram que os depósitos de lipofuscina na STGD tipo 3 tendem a ser um pouco maiores que na STGD tipo 1, e frequentemente assumem uma aparência de “borboleta” nas imediações das lesões maculares2. Os depósitos de lipofuscina eram de pequena dimensão em nossos três pacientes e não foi observado tal aspecto em “borboleta”.
REFERÊNCIAS
1. Kniazeva M, Chiang MF, Morgan B, Anduze AL, Zack DJ, Han M, et al. A new locus for autosomal dominant stargardt-like disease maps to chromosome 4. Am J Hum Genet. 1999;64(5):1394-9. http://dx.doi.org/10.1086/302377
2. Ryan SJ, Schachat AP, Wilkinson CP, Hinton DR, Sadda SR, Wiedemann P. Retina. 5th ed. London: Saunders; 2013.
3. Anmarkrud N. Fundus fluorescein angiography in fundus flavimaculatus and Stargardt’s disease. Acta Ophtalmol. 1979;57(2):172-82. Abstract disponível em: https://www.ncbi.nlm.nih.gov/pubmed/?term=Acta+Ophtalmol.+1979%3B57(2)%3A172-82
4. Ernest JT, Krill AE. Fluorescein studies angiography in fundus flavimaculatus and drusen. Am J Ophtalmol. 1966;62(1):1-6.
5. Lois N, Halfyard AS, Bird AC, Holder GE, Fitzke FW. Fundus autofluorescence in Stargardt macular dystrophy - fundus flavimaculatus. Am J Ophtalmol. 2004;138(1):55-63. http://dx.doi.org/10.1016/j.ajo.2004.02.056
6. Grandinetti AA, Portella E, Arana J, Iskorostenski NTV. Subretinal fibrosis in Stargardt’s disease: case report. Arq Bras Oftalmol 2011 ;74(6):449-51. http://dx.doi.org/10.1590/S0004-27492011000600015
7. Kaplan J, Gerber S, Larget-Piet D, Rozet JM, Dollfus H, Dufier JL, et al. A gene for Stargardt’s disease (fundus flavimaculatus) maps to the short arm of chromosome 1. Nat Genet. 1993;5:396-9. http://dx.doi.org/10.1038/ng1193-308
8. Zhang K, Bither PP, Park R, Donoso LA, Siedman JG, Siedman CE. A dominant Stargardt’s macular dystrophy maps to chromosome 13q34. Arch Ophthalmol. 1994;112:759-64. http://dx.doi.org/10.1001/archopht.1994.01090180057035
9 Stone EM, Nichols BE, Kimura AE, Weingeist TA, Drack A, Sheffield VC. Clinical features of a Stargardt-like dominant progressive macular dystrophy with genetic linkage to chromosome 6q. Arch Ophthalmol. 1994;112:765-72. http://dx.doi.org/10.1001/archopht.1994.01090180063036
10. Zhang K, Kniazeva M, Han M, Li W, Yu Z, Yang Z, et al. A 5-bp deletion in ELOVL4 is associated with two related forms of autosomal dominant macular dystrophy. Nat Genet. 2001 ;27(1):89-93. http://dx.doi.org/10.1038/83817
11. Bernstein PS, Tammur J, Singh N, Hutchinson A, Dixon M, Pappas CM, et al. Diverse macular dystrophy phenotype caused by a novel complex mutation in the ELOVL4 gene. Invest Ophthalmol Vis Sci. 2001; 42(13):3331-6. Disponível em: http://iovs.arvojournals.org/article.aspx?articleid=2123484
12. Unonius N, Farah ME, Sallum JMF. Classificação diagnóstica dos portadores de doenças degenerativas de retina integrantes dos grupos Retina São Paulo e Retina Vale do Paraíba. Arq Bras Oftalmol. 2003;66(4):443-8. http://dx.doi.org/10.1590/S0004-27492003000400009
13. Fishman GA. Fundus flavimaculatus: a clinical classification. Arch Ophtalmol. 1976;94:2061 -7. http://dx.doi.org/10.1001/archopht.1976.03910040721003
14. Edwards AO, Miedziak A, Vrabec T, Verhoeven J, Acott TS, Weleber RG, et al. Autosomal dominant stargardt-like macular dystrophy: clinical characterization, longitudinal follow-up, and evidence for a common ancestry in families linked to chromosome 6q14. Am J Ophthalmol. 1999;127(4):426-35. http://dx.doi.org/10.1016/S0002-9394(98)00331-6
15. Lopez PF, Maumenee IH, de la Cruz Z, Green WR. Autosomal-dominant fundus flavimaculatus: clinicopathologic correlation. Ophthalmology. 1990;97:798-809. http://dx.doi.org/10.1016/S0161-6420(90)32508-3
Fonte de financiamento: declaram não haver.
Conflito de interesses: declaram não haver.
Recebido em:
13 de Novembro de 2016.
Aceito em:
29 de Novembro de 2016.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados



 Ler em inglês
Ler em inglês
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket