

Felipe Di Domenico1; Felipe Kenzo Ishida2; Murilo Simão Cenovicz3
DOI: 10.17545/e-oftalmo.cbo/2016.73
ABSTRACT
Stargardt disease is a rare early-onset macular dystrophy, which progressively affects central visual acuity. In most cases, it is caused by an autosomal recessive inheritance. Here, we present the case of a family with Stargardt disease caused by autosomal dominant inheritance, together with a description of their history, clinical findings, and pertinent complementary examinations.
Keywords: Macular Degeneration: Retinal Cone Photoreceptor Cells: Macular Dystrophy: ELOVL4 protein, human
RESUMO
A doença de Stargardt é uma rara distrofia macular de início precoce que afeta progressivamente a acuidade visual central. Na maior parte das vezes é causada por uma herança autossômica recessiva. Apresentamos o caso de uma família com doença de Stargardt causada por herança autossômica dominante, descrevendo sua história, achados clínicos e exames complementares relevantes.
Palavras-chave: Degeneração Macular; Células Fotorreceptoras Retinianas Cones; Distrofia Macular; Proteína humana ELOVL4
INTRODUCTION
Stargardt disease (STGD) is the most common recessive macular dystrophy, and is characterized by decreased central vision, atrophy of the macula and the underlying retinal pigment epithelium, and frequent presence of yellowish flecks in the retinal posterior pole1.
Clinical manifestations of the disease vary depending on its genotype, as well as on the sensitivity of the foveal cones and the retinal pigmented epithelium (RPE). Low visual acuity is the most common clinical manifestation, and may vary from 20/30 to 20/200 whereas age of onset can vary from 5 to 50 years2.
Clinical manifestations observed in the fundus include sparse yellowish flecks, which reflect the accumulation of bisretinoids underlying the RPE, and atrophy of the RPE in the macular region, which is often, described as resembling “beaten bronze”2. Examination by fluorescein angiography (FA) shows that bisretinoids that have accumulated adjacent to the choroid can block light excitation of the dye in the choroidal circulation, which is known as “choroidal silence,” thus leaving the retinal vessels exposed in a deep hypofluorescence a4. Other signs include RPE atrophy, “bull's eye” maculopathy, hyperfluorescent flecks, and preservation of peridiscal RPE (peripapillary sparing)5.
Genetically, STGD is a heterogeneous disorder, mostly inherited as an autosomal recessive disorder, and very rarely as an autosomal dominant trait with a late onset of clinical symptoms 6. A locus corresponding to a recessive allele was mapped on chromosome 1 p (STGD Type 1)7. This gene encodes a retina-specific transmembrane protein, ABCA4, which belongs to the ATP Binding Cassette (ABC) membrane transporter group 1 Few cases of families diagnosed with Stargardt disease related to inheritance of an autosomal dominant trait were described, such as the mapping of a locus on chromosome 13q (STGD type 2)8, chromosome 6q (STGD Type 3)9, and chromosome 4p (STGD Type 4) 1.
Dominant Stargardt-like macular dystrophy (Stargardt disease Type 3) was first described by Stone et al., in 1994, in large family that exhibited a phenotype that was similar to Stargardt disease that then led to the mapping of the gene on chromosome 6q9. In 2011, Zhang et al. identified a deletion of 5 base pairs from the ELOVL4 (Elongation Of Very Long Chain Fatty Acids) gene on chromosome 6q in five families affected by this disease 10. Conversely, also in 2011, Bernstein et al. identified a complex heterozygous mutation in the ELOVL4 gene, wherein two deletions of a single base pair each, separated by 4 nucleotides, resulted in a frameshift in the gene and consequent truncation of the protein ELOVL4, indicating that the phenotype resulting from these mutations varies greatly among patients11.
The ELOVL4 gene encodes an enzyme in the endoplasmic reticulum responsible for the biosynthesis of fatty acids that contain more than 26 carbon atoms. This enzyme is expressed in the brain and retina, where its expression is restricted to photoreceptor cells. Mutations associated with STGD Type 3 compromise the mutated enzyme's trafficking, which results in an accumulation of lipofuscin, leading to death of photoreceptor cells2.
The clinical manifestations of STGD Type 3 are very similar to those found in STGD Type 1. Deposits of lipofuscin in STGD Type 3 tend to be somewhat larger than the ones observed on STGD Type 1, and they often exhibit a “butterfly” appearance near macular injuries2. The RPE is usually normal, and examination by fluorescein angiography shows a lower prevalence of silent choroid 2.
Here, we report the case of a family diagnosed with Stargardt disease Type 3, together with a description of their history, clinical findings, and pertinent complementary examinations.
CASE REPORT
PATIENT 1
Female, 47 years old. Main complaint: “blurry vision.”
Current medical history: The patient reported a progressive worsening of her visual acuity throughout her life, but it had become worse in the last 3 years. She had symptoms in both eyes, with greater intensity in the right eye. The patient reported that doctors from another clinic suspected retinal dystrophy approximately 10 years earlier, but no further investigation was done. The patient did not have a history of systemic diseases, surgeries, hospitalizations, nor allergies.
Family history: The patient reported that her mother, siblings, and daughter were suspected to have retinal dystrophy.
Physical Examination:
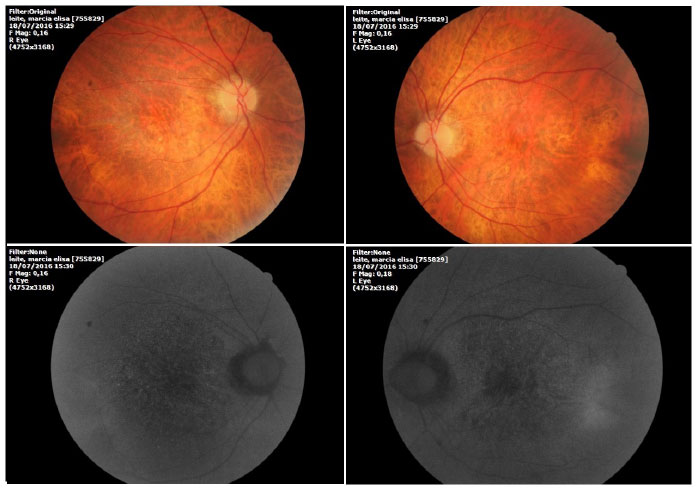
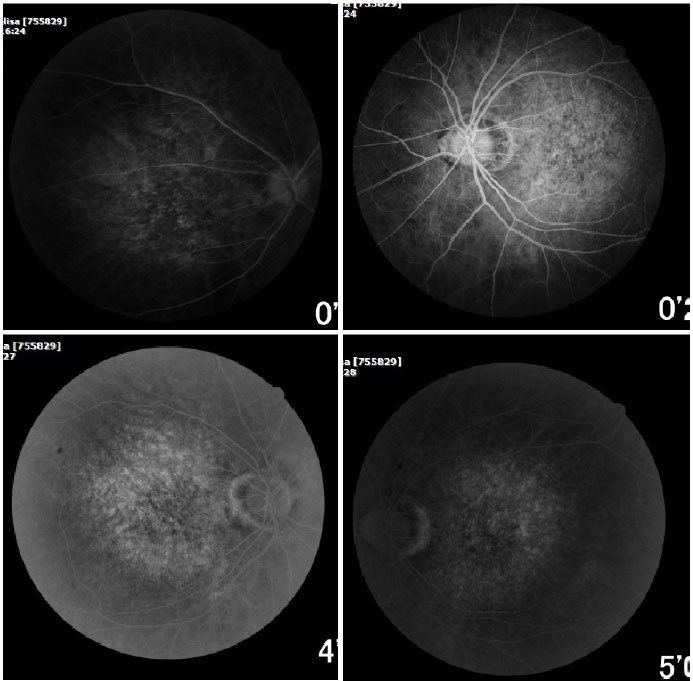
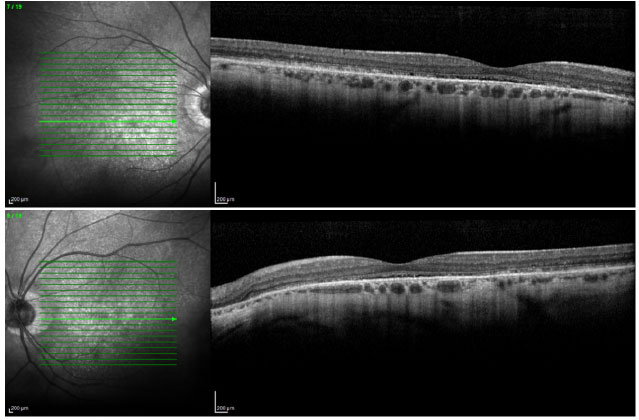
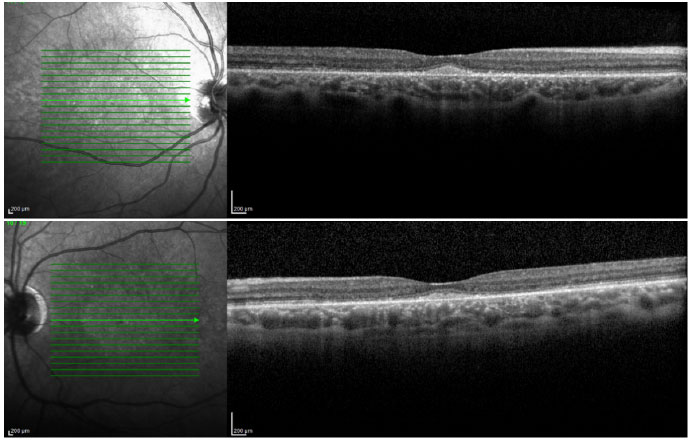
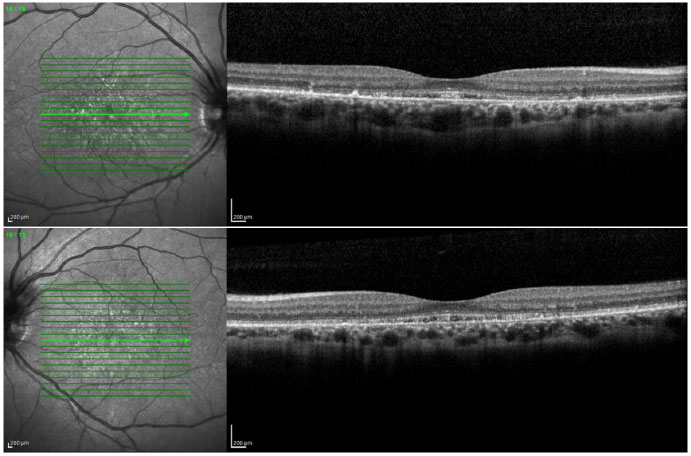
Ophthalmic examination showed a visual acuity of 20/400 in the right eye (RE) and 20/200 in the left eye (LE), both with the best optical correction. Results of biomicroscopy showed no changes, and the tonometry test measured pressure of 15 mmHg in both eyes (BE). Fundoscopic examination showed diffuse atrophy of the RPE with the presence of sparse hazy flecks in both eyes (Figure 1). Fluorescein angiography showed hyperfluorescence due to a window defect of the macular and perimacular area, exhibiting a coalescent aspect of hyperfluorescent dots up to the late phases (Figure 2). Optical coherence tomography revealed changes in the ellipsoid layer with loss of photoreceptors (Figure 3).
FAMILY INVESTIGATION
PATIENT 2
The brother (male, age 39 years) of Patient 1 came to our hospital. He brought his electroretinogram results showing a photopic change. When questioned about his eye symptoms, the patient described intense photophobia. Eye examination showed a visual acuity of 20/30 in the RE and 20/80 in LE, with the best optical correction in BE. Results of biomicroscopy showed no changes, and the tonometry test measured 12 mmHg pressure in BE.
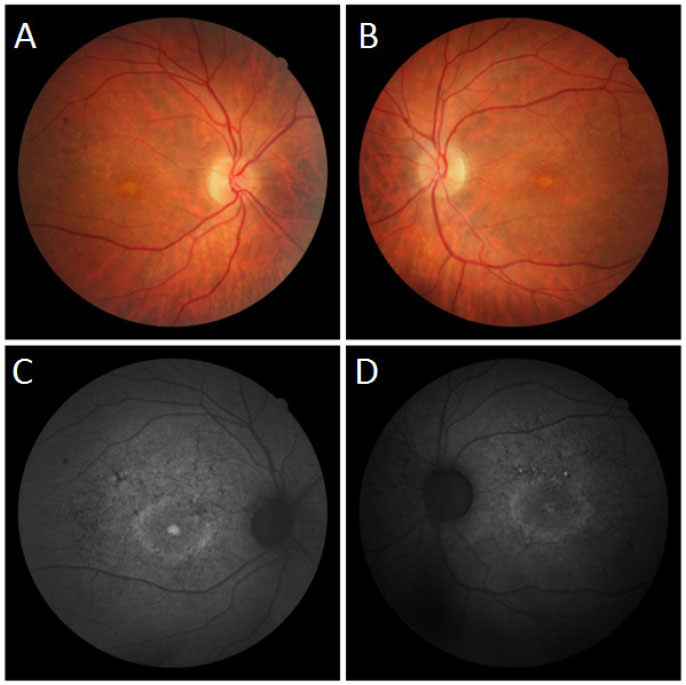
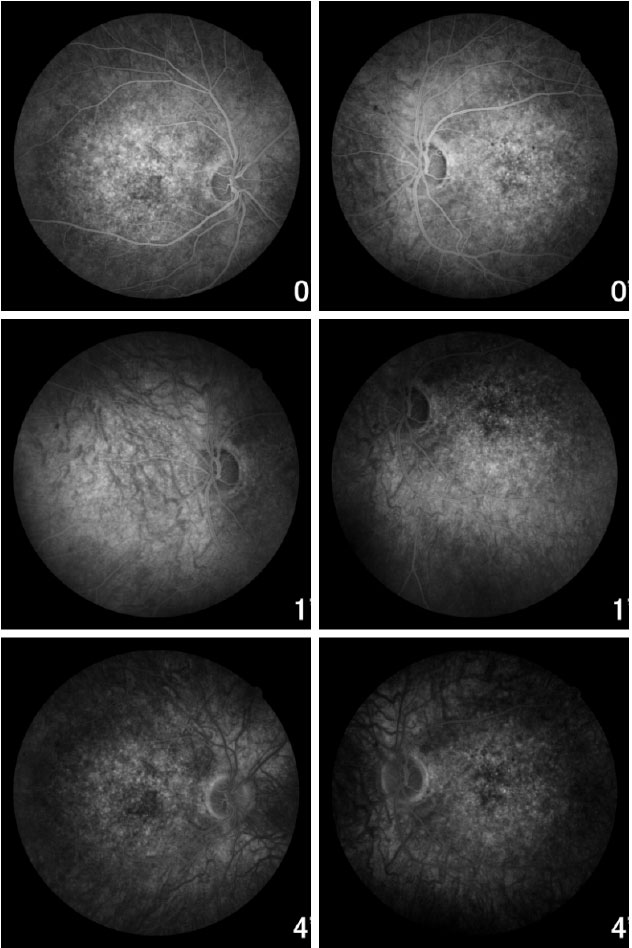
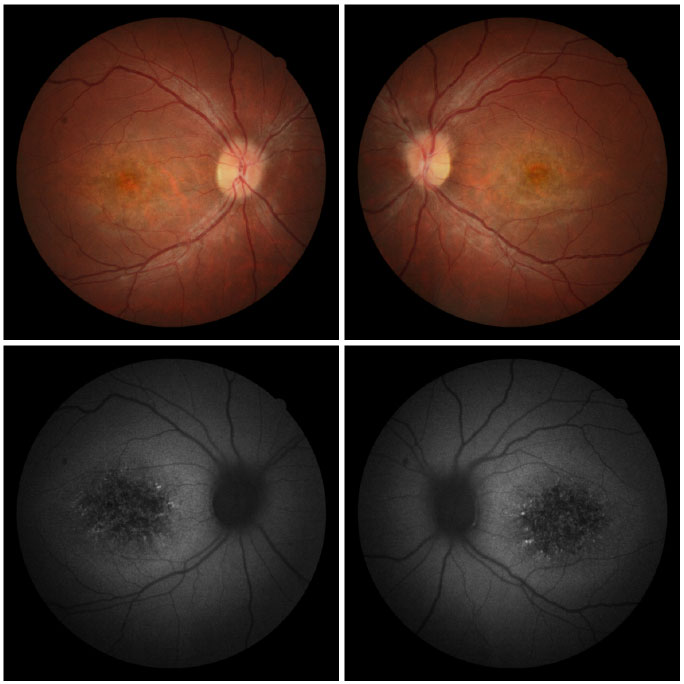
Fundoscopic evaluation revealed a diffuse atrophy of the RPE, in addition to a macula with beaten bronze appearance and the presence of punctiform flecks in the perimacular region in both eyes (Figure 4 A-B). On imaging with red-free light filter, bull's eye maculopathy was observed, with perimacular and foveal hyperautofluorescent areas as well as punctiform atrophies in the central RPE (Figure 4 C-D). Fluorescein angiography showed hyperfluorescence due to a window defect of the macular and perimacular area, exhibiting a coalescent aspect of hyperfluorescent dots up to the late phases (Figure 5). Optical coherence tomography revealed foveal deposits and changes in the ellipsoid layer of photoreceptors (Figure 6).
FAMILY INVESTIGATION
PATIENT 3
The daughter (female, age 29 years) of Patient 1 came to the hospital. She had no ocular symptoms, but reported a prior diagnosis of systemic lupus erythematosus and polyarteritis nodosa. In addition, she had suffered traumatic rupture of a knee ligament 3 weeks earlier. Optometry revealed a visual acuity of 20/20 in the RE and 20/25 in the LE, with the best optical correction in BE. Results of biomicroscopy showed no changes, and tonometry test measured 12 mmHg pressure in BE. Fundoscopic evaluation revealed a beaten bronze appearance in the macular region, atrophy of the RPE in the central retinal region, and the presence of sparse flecks in the perimacular region in BE (Figure 7).
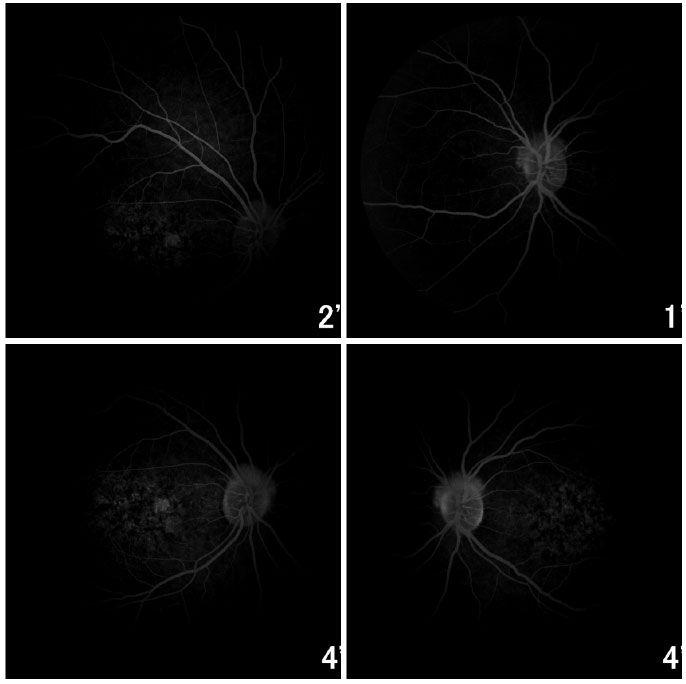
Fluorescein angiography showed an intense choroidal silence, with contrast between the retinal vessels and the dark background of the choroid. There were dotted hyperfluorescent areas caused by a window defect in the macular and perimacular area in BE (Figure 8). Optical coherence tomography revealed deposits underneath the RPE and changes in the ellipsoid layer (Figure 9).
DISCUSSION
The recessive form of Stargardt disease is the most common hereditary macular dystrophy, representing 7% of retinal dystrophies 12. In contrast, Stargardt-like macular dystrophy (STGD Type 3) has been reported only in a few cases in the literature, being an atypical disease even in specialized medical centers. According to Fishman, the initial diagnosis of recessive Stargardt disease is more prevalent in the age range 20-2913. Studies did not report any data pertaining to age of the initial diagnosis in patients with autosomal dominant Stargardt disease. Our 3 patients, aged 47, 39, and 29 years, were only diagnosed in our clinic.
STGD patients frequently experience photophobia, and they seem to have better night vision14, which was reported by Patient 2. Our patients presented with normal color vision, which was to be expected according to reports in the literature. Usually, altered color vision is not observed in patients with dominant Stargardt disease14.
Edwards et al. found visual acuity in Stargardt Type 3 patients ranging from 20/20 to 20/800, and most patients older than 30 years presented with vision 20/200 or worse 14. After reaching a visual acuity of 20/200, progression is slow, which is different from that in recessive Stargardt disease. Surprisingly, our findings showed the opposite: Patient 2, 39 years old, presented with a visual acuity of 20/30 in the RE and 20/80 in the LE, and Patient 3, 29 years old, presented with a visual acuity of 20/20 in the RE and 20/25 in the LE, whereas Patient 1, 47 years old, presented with a visual acuity of 20/200 in the RE and 20/400 in LE, as reported in the literature.
Edwards et al. studied two families with Stargardt disease Type 3, and found patterns for changes observed in the fundoscopic examination according to disease stage14. Initial findings included sub-foveal atrophy of the RPE, associated with an area of window defect of the macula on fluorescein angiography. Over the years, there would be an increased atrophy of the RPE that resembles the stained lesions observed in patients with recessive Stargardt disease. Flecks were generally absent at that stage. Over several years, in most patients, injuries have evolved to a well-circumscribed retinal area, RPE atrophy, and a choroid of increasing size that is associated with flecks. The atrophic lesion often had a beaten bronze appearance. We did not have the opportunity to conduct a horizontal follow-up of patients at the time of the study. However, fundoscopic changes found in our three patients match the findings described by Edwards et al. 14
Families with autosomal dominant Stargardt macular dystrophy linked to chromosomes 6q14 and 13q34 display a similar fundus phenotype 14. According to Edwards et al., families linked to 6q14 do not have the dark choroid observed in recessive Stargardt disease, and it is not known whether the families linked to 13q34 exhibit choroidal silence. Lopez et al. described a family with an autosomal dominant Stargardt macular dystrophy that presented choroidal silence15. In our patients, fluorescein angiography detected choroidal silence in Patient 3 (Figure 8). Ryan et al. reported that deposits of lipofuscin in STGD Type 3 tend to be somewhat larger than in STGD Type 1, and often have the appearance of a butterfly near the macular injuries2. The lipofuscin deposits were small in our three patients, and they did not have a butterfly appearance.
REFERENCES
1. Kniazeva M, Chiang MF, Morgan B, Anduze AL, Zack DJ, Han M, et al. A new locus for autosomal dominant stargardt-like disease maps to chromosome 4. Am J Hum Genet. 1999;64(5):1394-9. http://dx.doi.org/10.1086/302377
2. Ryan SJ, Schachat AP, Wilkinson CP, Hinton DR, Sadda SR, Wiedemann P. Retina. 5th ed. London: Saunders; 2013.
3. Anmarkrud N. Fundus fluorescein angiography in fundus flavimaculatus and Stargardt's disease. Acta Ophtalmol. 1979;57(2):172-82. Abstract disponível em: https://www.ncbi.nlm.nih.gov/pubmed/?term=Acta+Ophtalmol.+1979%3B57(2)%3A172-82
4. Ernest JT, Krill AE. Fluorescein studies angiography in fundus flavimaculatus and drusen. Am J Ophtalmol. 1966;62(1):1-6.
5. Lois N, Halfyard AS, Bird AC, Holder GE, Fitzke FW. Fundus autofluorescence in Stargardt macular dystrophy - fundus flavimaculatus. Am J Ophtalmol. 2004;138(1):55-63. http://dx.doi.org/10.1016/j.ajo.2004.02.056
6. Grandinetti AA, Portella E, Arana J, Iskorostenski NTV. Subretinal fibrosis in Stargardt's disease: case report. Arq Bras Oftalmol 2011 ;74(6):449-51. http://dx.doi.org/10.1590/S0004-27492011000600015
7. Kaplan J, Gerber S, Larget-Piet D, Rozet JM, Dollfus H, Dufier JL, et al. A gene for Stargardt's disease (fundus flavimaculatus) maps to the short arm of chromosome 1. Nat Genet. 1993;5:396-9. http://dx.doi.org/10.1038/ng1193-308
8. Zhang K, Bither PP, Park R, Donoso LA, Siedman JG, Siedman CE. A dominant Stargardt's macular dystrophy maps to chromosome 13q34. Arch Ophthalmol. 1994;112:759-64. http://dx.doi.org/10.1001/archopht.1994.01090180057035
9 Stone EM, Nichols BE, Kimura AE, Weingeist TA, Drack A, Sheffield VC. Clinical features of a Stargardt-like dominant progressive macular dystrophy with genetic linkage to chromosome 6q. Arch Ophthalmol. 1994;112:765-72. http://dx.doi.org/10.1001/archopht.1994.01090180063036
10. Zhang K, Kniazeva M, Han M, Li W, Yu Z, Yang Z, et al. A 5-bp deletion in ELOVL4 is associated with two related forms of autosomal dominant macular dystrophy. Nat Genet. 2001 ;27(1):89-93. http://dx.doi.org/10.1038/83817
11. Bernstein PS, Tammur J, Singh N, Hutchinson A, Dixon M, Pappas CM, et al. Diverse macular dystrophy phenotype caused by a novel complex mutation in the ELOVL4 gene. Invest Ophthalmol Vis Sci. 2001; 42(13):3331-6. Disponível em: http://iovs.arvojournals.org/article.aspx?articleid=2123484
12. Unonius N, Farah ME, Sallum JMF. Classificação diagnóstica dos portadores de doenças degenerativas de retina integrantes dos grupos Retina São Paulo e Retina Vale do Paraíba. Arq Bras Oftalmol. 2003;66(4):443-8. http://dx.doi.org/10.1590/S0004-27492003000400009
13. Fishman GA. Fundus flavimaculatus: a clinical classification. Arch Ophtalmol. 1976;94:2061 -7. http://dx.doi.org/10.1001/archopht.1976.03910040721003
14. Edwards AO, Miedziak A, Vrabec T, Verhoeven J, Acott TS, Weleber RG, et al. Autosomal dominant stargardt-like macular dystrophy: clinical characterization, longitudinal follow-up, and evidence for a common ancestry in families linked to chromosome 6q14. Am J Ophthalmol. 1999;127(4):426-35. http://dx.doi.org/10.1016/S0002-9394(98)00331-6
15. Lopez PF, Maumenee IH, de la Cruz Z, Green WR. Autosomal-dominant fundus flavimaculatus: clinicopathologic correlation. Ophthalmology. 1990;97:798-809. http://dx.doi.org/10.1016/S0161-6420(90)32508-3
Funding source: None
Conflicts of interest: None
Received on:
November 13, 2016.
Accepted on:
November 29, 2016.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados



 Ler em português
Ler em português
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket