

Vitor Yuzo Inada1; Victor de Oliveira Campos1; Mariana Matioli da Palma1,2
DOI: 10.17545/eOftalmo/2023.0011
RESUMO
A Retinose Pigmentar é uma distrofia hereditária da retina, rara, que pode levar a uma importante baixa visual. Acomete primeiramente os bastonetes e com o tempo pode acometer os cones. Esta doença apresenta diferentes tipos de herança: autossômica dominante, autossômica recessiva (forma mais comum) e ligada ao X. Este relato de caso tem como objetivo apresentar o caso de uma paciente com Retinose Pigmentar autossômica dominante associada a uma variante patogênica c.166G>A(p.Gly56Arg) no gene NR2E3.
Palavras-chave: Retinite Pigmentosa; NR2E3; Síndrome do Cone-S.
ABSTRACT
Retinitis Pigmentosa is a rare hereditary retinal dystrophy that can lead to significant low vision. It primarily affects the rod phtoreceptors and later it can affect the cones. This disease has different types of inheritance: autossomal dominant, autossomal recessive (most commom form) and X-linked. This case report aims to present the case of a patient with autossomal dominant Retinitis Pigmentosa associated with a pathogenic variant c.166G>A(p.Gly56Arg) in the NR2E3 gene.
Keywords: Retinitis Pigmentosa; NR2E3; Enhanced S-Cone Syndrome.
INTRODUÇÃO
A retinose pigmentar (RP) apresenta uma tríade clássica de achados retinianos composta pela presença de espículas ósseas, palidez de disco e atenuação vascular1. Apresenta diferentes tipos de padrão de herança: autossômica dominante, autossômica recessiva (forma mais comum), ligado ao X e esporádica2. A RP, é uma distrofia hereditária da retina, progressiva que afeta aproximadamente 1 a cada 4.000 individuos, ela é considerada uma doença rara3. Cerca de 69 genes já foram associados a RP4. Muitos desses genes têm estudos clínicos em andamento, tornando essencial o diagnóstico molecular preciso.
RELATO DE CASO
Paciente de 57 anos, sexo feminino, compareceu a consulta oftalmológica em um serviço público com queixa de baixa acuidade visual progressiva há cerca de 2 anos. Relatava que teve diagnóstico de RP aos 43 anos de idade em outro serviço, mas perdeu o seguimento.
De história patológica pregressa paciente apresentava hipertensão arterial sistêmica, insuficiência cardíaca e era tabagista.
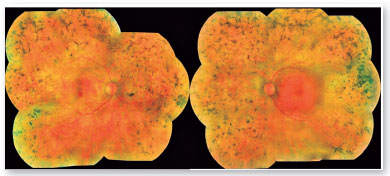
Ao exame oftalmológico, a melhor acuidade visual corrigida era contagem de dedos em olho direito e 20/40 em olho esquerdo. À biomicroscopia não foram evidenciadas alterações no segmento anterior e a pressão intraocular estava 16 mmHg em olho direito e 15 mmHg em olho esquerdo. Ao mapeamento de retina foram detectadas as seguintes alterações nos dois olhos: disco óptico pálido, atenuação e afinamento vascular e espículas ósseas em toda a periferia 360º (Figura 1).
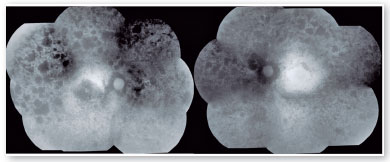
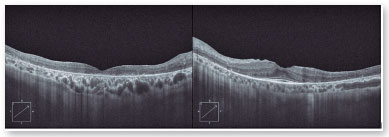
O exame de autofluorescência demonstrou lesões hipoautofluorescentes em média periferia e na periferia em ambos os olhos. O olho direito apresentava lesões hipoautofluorescentes em região macular (Figura 2). O exame de tomografia de coerência óptica demonstrou atrofia da retina externa e presença da zona elipsoide apenas na região foveal de olho esquerdo. Seu olho direito apresenta atrofia da zona elipsoide na área foveal, o que justificava sua pior acuidade visual em olho direito (Figura 3).
O resultado do teste genético identificou uma variante c.166G>A(p.Gly56Arg) no gene NR2E3, classificada como patogênica5, que está associada diretamente a casos autossômicos dominantes da doença. A presença do aminoácido glicina na posição 56 da proteína é altamente conservada ao longo de diferentes espécies e essa mutação afeta provavelmente a função ou a estrutura da proteína6 sendo então considerada como causadora da doença. A paciente relatava ter 4 primos com retinose pigmentar e que sua avó materna apresentava dificuldade visual, sem diagnóstico definido. Paciente negava ter filhos e sua mãe já era falecida.
DISCUSSÃO
Variantes patogênicas no gene receptor nuclear, NR2E3 estão associadas à síndrome do cone S autossômico recessivo (ESCS), síndrome de Goldman-Favre, degeneração retiniana pigmentar (CPRD), retinose pigmentar autossômica recessiva e retinose pigmentar autossômica dominante. De acordo com a base de dados de mutações genéticas humanas, HGMD [http://www.hgmd.cf.ac.uk/ac/] mais de 75 variantes já foram descritas no gene NR2E3. A maioria das variantes são mutações missense e a maioria está associada a ESCS.
A variante patogênica c.166G>A(p.Gly56Arg) encontrada na paciente brasileira já foi descrita na literatura como causadora de retinose pigmentar autossômica dominante. Em uma coorte de 24 pacientes com retinose pigmentar associada a mesma variante da paciente do estudo, a média de aparecimento de nictalopia foi aos 15 anos de idade e a média do diagnóstico foi aos 30 anos de idade4, diferente da nossa paciente que teve um diagnóstico mais tardio. A nictalopia é o sintoma mais frequente entre os pacientes de retinose pigmentar.
Escher e cols. reportaram a presença de um duplo anel hiperautofluorescente associado a variante descrita7. Esse achado não foi considerado patognomônico e não foi encontrado na paciente do estudo, que apresentava um fenótipo mais avançado da doença com uma distrofia difusa da retina.
Ainda não há tratamento aprovado para retinose pigmentar autossômica dominante. Todavia, há um estudo clínico em andamento para avaliar a segurança e eficácia de terapia gênica para pacientes com retinose pigmentar associada ao gene NR2E38, demonstrando a importância do teste genético nos dias de hoje.
REFERÊNCIAS
1. Kanski JJ. Distrofias Retinianas. In: Kanski JJ. Oftalmologia Clínica: Uma Abordagem Sistêmica. 6ª. edição. Rio de Janeiro: Editora Elsevier; 2008; 663-67.
2. Queiroz ACC, Frasson M, Veloso CER, Arantes RR, Nehemy MB. Estudo clínico e padrão de herança em pacientes com retinose pigmentar. Rev Bras Oftalmol. 2013;72(1).
3. Fahim A. Retinitis pigmentosa: recent advances and future directions in diagnosis and management. Curr Opin Pediatr. 2018;30(6):725-733.
4. Blanco-Kelly F, García-Hoyos M, Lopez-Martinez MA, Lopez-Molina MI, Riveiro-Alvarez R, Fernandez-San Jose P, et al. Dominant Retinitis Pigmentosa, p.Gly56Arg Mutation in NR2E3: Phenotype in a Large Cohort of 24 Cases. PLoS One. 2016 Feb 24;11(2):e0149473.
5. National Center for Biotechnology Information. ClinVar. Cited 2022 Dec 13,2022]. Available from: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV001076269.4
6. Coppieters F, Leroy BP, Beysen D, Hellemans J, De Bosscher K, Haegeman G, et al. Recurrent mutation in the first zinc finger of the orphan nuclear receptor NR2E3 causes autosomal dominant retinitis pigmentosa. Am J Hum Genet. 2007;81(1):147-57.
7. Escher P, Tran HV, Vaclavik V, Borruat FX, Schorderet DF, Munier FL. Double concentric autofluorescence ring in NR2E3-p.G56R-linked autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2012;53(8):4754-64.
8. Clinical Trials [Internet]. Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information. [cited 2022 Nov 25]. Available from: https://clinicaltrials.gov/ct2/show/NCT05203939?cond=NR2E3&draw=2&rank=1



Financiamento: Declaram não haver.
Conflitos de Interesse: Declaram não haver.
Recebido em:
9 de Dezembro de 2022.
Aceito em:
10 de Janeiro de 2023.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados
 Ler em inglês
Ler em inglês
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket