

Vitor Yuzo Inada1; Victor de Oliveira Campos1; Mariana Matioli da Palma1,2
DOI: 10.17545/eOftalmo/2023.0011
ABSTRACT
Retinitis Pigmentosa is a rare hereditary retinal dystrophy that can lead to significant low vision. It primarily affects the rod phtoreceptors and later it can affect the cones. This disease has different types of inheritance: autossomal dominant, autossomal recessive (most commom form) and X-linked. This case report aims to present the case of a patient with autossomal dominant Retinitis Pigmentosa associated with a pathogenic variant c.166G>A(p.Gly56Arg) in the NR2E3 gene.
Keywords: Retinitis Pigmentosa; NR2E3; Enhanced S-Cone Syndrome.
RESUMO
A Retinose Pigmentar é uma distrofia hereditária da retina, rara, que pode levar a uma importante baixa visual. Acomete primeiramente os bastonetes e com o tempo pode acometer os cones. Esta doença apresenta diferentes tipos de herança: autossômica dominante, autossômica recessiva (forma mais comum) e ligada ao X. Este relato de caso tem como objetivo apresentar o caso de uma paciente com Retinose Pigmentar autossômica dominante associada a uma variante patogênica c.166G>A(p.Gly56Arg) no gene NR2E3.
Palavras-chave: Retinite Pigmentosa; NR2E3; Síndrome do Cone-S.
INTRODUCTION
Retinitis pigmentosa (RP) presents a classic triad of retinal findings composed of the presence of bony spicules, disc pallor, and vascular attenuation1. It presents different types of inheritance patterns: autosomal dominant, autosomal recessive (most common form), and sporadic X-linked2. RP, a progressive hereditary retinal dystrophy that affects 1 in 4,000 people, is a rare disease3. About 69 genes have already been associated with RP4. Many of these genes have ongoing clinical studies, making an accurate molecular diagnosis essential.
CASE REPORT
A 57-year-old female patient sought ophthalmological consultation from a public service after complaining of low progressive visual acuity for the past two years. She reported that she was diagnosed with RP at 43 years of age in another service, but was lost to follow-up.
With a previous pathological history, the patient presented with systemic arterial hypertension, heart failure, and was a smoker.
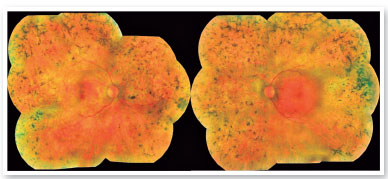
On ophthalmological examination, the best corrected visual acuity was classified using the scale “counting fingers” in the right eye and 20/40 in the left eye. On biomicroscopy, there were no alterations in the anterior segment and intraocular pressure was 16 mmHg in the right eye and 15 mmHg in the left eye. Retinal mapping detected the following changes in both eyes: pale optic disc, retinal vascular attenuation and thinning, and bone spicules all over the 360º peripheral retina (Figure 1).
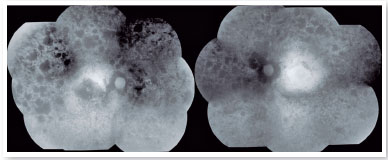
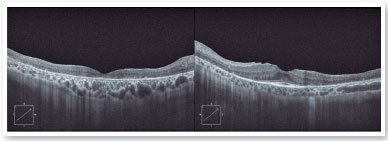
The autofluorescence test showed hypoautofluorescent lesions in the mid-periphery and in the periphery of the retina in both eyes. The right eye presented with hypoautofluorescent lesions in the macular region (Figure 2). Optical coherence tomography examination showed outer retinal atrophy and the presence of the ellipsoid zone only in the foveal region of the left eye retina. Her right eye presented ellipsoid zone atrophy in the foveal area, which justified her worse visual acuity in the right eye (Figure 3).
The results of the genetic testing identified a variant c.166G>A (p.Gly56Arg) in the NR2E3 gene, classified as pathogenic5, which is directly associated with autosomal dominant disease cases. The presence of the glycine amino acid at position 56 of the protein is highly conserved throughout different species and this mutation probably affects protein function or structure6 and is then considered the cause of the disease. The patient reported having four cousins with RP and that her maternal grandmother presented with visual difficulty, with no defined diagnosis. The patient denied having children and her mother was already dead.
DISCUSSION
Pathogenic variants in the nuclear receptor gene, NR2E3, are associated with the autosomal recessive inherited retinal S-cone syndrome (ESCS), Goldman-Favre syndrome, pigmentary retinal degeneration (CPRD), autosomal recessive RP, and autosomal dominant RP. According to the human genetic mutation database, HGMD [http://www.hgmd.cf.ac.uk/ac/] more than 75 variants have already been described in the NR2E3 gene. Most variants are missense mutations and are associated with ESCS.
The pathogenic variant c.166G>A (p.Gly56Arg) found in the Brazilian patient has been described in the literature as causing autosomal dominant RP. In a cohort study of 24 patients with RP associated with the same variant of the patient in the study, the average onset of nyctalopia symptoms was at 15 years of age and the average age of diagnosis was 30 years4, different from our patient who had a later diagnosis. Nyctalopia is the most frequent symptom among patients with RP.
Escher et al. reported the presence of a double hyperautofluorescent ring associated with the described variant7. This finding was not considered pathognomonic and was not found in the study patient, who presented a more advanced phenotyping of the disease with diffuse retinal dystrophy.
For autosomal dominant RP, there is currently no authorized treatment. However, a clinical trial evaluating the safety and efficacy of gene therapy for patients with RP linked to the NR2E38 gene is under underway, highlighting the significance of genetic testing nowadays.
REFERENCES
1. Kanski JJ. Distrofias Retinianas. In: Kanski JJ. Oftalmologia Clínica: Uma Abordagem Sistêmica. 6ª. edição. Rio de Janeiro: Editora Elsevier; 2008; 663-67.
2. Queiroz ACC, Frasson M, Veloso CER, Arantes RR, Nehemy MB. Estudo clínico e padrão de herança em pacientes com retinose pigmentar. Rev Bras Oftalmol. 2013;72(1).
3. Fahim A. Retinitis pigmentosa: recent advances and future directions in diagnosis and management. Curr Opin Pediatr. 2018;30(6):725-733.
4. Blanco-Kelly F, García-Hoyos M, Lopez-Martinez MA, Lopez-Molina MI, Riveiro-Alvarez R, Fernandez-San Jose P, et al. Dominant Retinitis Pigmentosa, p.Gly56Arg Mutation in NR2E3: Phenotype in a Large Cohort of 24 Cases. PLoS One. 2016 Feb 24;11(2):e0149473.
5. National Center for Biotechnology Information. ClinVar. Cited 2022 Dec 13,2022]. Available from: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV001076269.4
6. Coppieters F, Leroy BP, Beysen D, Hellemans J, De Bosscher K, Haegeman G, et al. Recurrent mutation in the first zinc finger of the orphan nuclear receptor NR2E3 causes autosomal dominant retinitis pigmentosa. Am J Hum Genet. 2007;81(1):147-57.
7. Escher P, Tran HV, Vaclavik V, Borruat FX, Schorderet DF, Munier FL. Double concentric autofluorescence ring in NR2E3-p.G56R-linked autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2012;53(8):4754-64.
8. Clinical Trials [Internet]. Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information. [cited 2022 Nov 25]. Available from: https://clinicaltrials.gov/ct2/show/NCT05203939?cond=NR2E3&draw=2&rank=1



Funding: No specific financial support was available for this study.
Conflict of interest: None of the authors have any potential conflict of interest to disclose.
Received on:
December 9, 2022.
Accepted on:
January 10, 2023.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados
 Ler em português
Ler em português
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket