

João Marçal Medeiros de Sousa1; Clayton Leite de Moura1; Hanniman Denizard Cosme Barbosa1; Thales Matheus Lopes de Sousa1; Lisley Medeiros Garcia1; Gabriela de Araújo Miranda1; Aganeide Castilho Palitot2
DOI: 10.17545/eOftalmo/2022.0002
ABSTRACT
OBJECTIVES: To assess whether systemic and localized sclerosis have a significant correlation with neuromyelitis optica and its spectrum of diseases, aiming to open new study perspectives for these autoimmune diseases.
METHODS: This is an integrative review of the literature of the last 20 years, which aims to investigate the relationship between neuromyelitis optica, systemic scleroderma, and localized scleroderma. Articles in Portuguese, English, and Spanish from the MEDLINE, LILACS, SciELO, and Scopus databases were searched using the keywords “neuromyelitis optica,” “systemic scleroderma,” and “localized scleroderma,” along with their synonyms and correspondents in Portuguese and Spanish.
RESULTS: The search generated 23 articles. Three case reports, one in English and two in Portuguese, comprised the final corpus of this work.
DISCUSSION: The relationship between neuromyelitis optica and autoimmune diseases is well established, but its association with systemic and localized scleroderma is rarely reported in the literature. Furthermore, the reported associations primarily arise from isolated cases, thereby making this association relatively rare. Uncommon testing, the subclinical or initial status of some neuromyelitis optica presentations, and immunosuppressive therapies may explain this rare association.
CONCLUSION: Active search and measurement studies of anti-aquaporin 4 in patients with autoimmune disorders are necessary. This is a large field of research that requires further investigation.
Keywords: Neuromyelitis optica; Systemic scleroderma; Localized scleroderma; aquaporin 4; Antibody.
RESUMO
OBJETIVOS: Neuromielite óptica é uma doença imunomediada cujo diagnóstico baseia-se na detecção do anticorpo anti-aquaporina 4. A neuromielite óptica é reconhecidamente associada a uma gama de doenças autoimunes.
MÉTODOS: Trata-se de uma revisão integrativa da literatura dos últimos 20 anos que busca investigar a relação entre a neuromielite óptica e o escleroderma sistêmico e a esclerodermia localizada. Incluiu-se artigos em português, inglês ou espanhol das bases de dados MEDLINE, Lilacs, SciELO e Scopus encontrados através das palavras-chave “Neuromielite óptica”, “Escleroderma Sistêmico” e “Esclerodermia Localizada”, seus sinônimos e correspondentes em inglês e espanhol.
RESULTADOS: A busca gerou 23 artigos. Três relatos de caso, um em inglês e dois e português, compuseram o corpus final deste trabalho.
DISCUSSÃO: A relação entre Neuromielite óptica e doenças autoimunes é estabelecida. Em relação ao escleroderma sistêmico e a esclerodermia localizada há pouca associação dentro da literatura, maioria oriunda de casos isolados, dando a esta associação o status de raridade. A baixa testagem, status sub-clínicos/iniciais de algumas apresentações da neuromielite óptica e terapias imunossupressoras podem explicar esta baixa associação.
CONCLUSÃO: Estudos de busca ativa e mensuração do anti-aquoporina 4 em pacientes com afecções auto-imunes se fazem necessários. Trata-se de um grande campo de pesquisa ainda a ser desbravado.
Palavras-chave: Neuromielite óptica; Escleroderma sistêmico; Esclerodermia localizada; Aquaporina 4; Anticorpo.
INTRODUCTION
Neuromyelitis optica (NMO) is an inflammatory autoimmune demyelinating disease with severe clinical repercussions(1). Its lesions are predominantly in the optic nerve and spinal cord, normally sparing the brain, unlike multiple sclerosis(2), of which NMO was once considered a localized variant(1). Until 2004, the diagnosis of NMO was based on minor, major, and paraclinical criteria(3), but the discovery of an antibody against the aquaporin 4 (AQP4) water channel protein (NMO–IgG) changed the approach to the disease as follows: immunoglobulin was consequently considered as its main etiologic agent, with high specificity and sensitivity(4), which made early diagnosis possible and provided better treatment and prognosis for patients with NMO(1).
AQP4 is the main water channel protein of the central nervous system (CNS) and is located in astrocytes. It is responsible for brain fluid balance and control of serum potassium and glutamate levels(2,5,6). The binding of anti-aquaporin 4 antibodies leads to a downregulation of these channels and causes astrocytic and, consequently, neuronal damage. In addition, it is responsible for generating inflammatory processes via complement and cellular immunity, demyelination, and edema in the CNS(6,7). Furthermore, it is argued that NMO-IgG increases the permeability of the blood-brain barrier, allowing antibodies, granulocytes, and T-cell-specific antigens to enter this normally inaccessible environment(8).
Notable, the discovery of NMO–IgG has opened a new field of study, as the same antibody was identified in other autoimmune diseases, thereby creating the concept of NMO Spectrum Disorders (NMOSD)(1,9). NMOSD include myasthenia gravis, hypothyroidism, pernicious anemia, scleroderma, ulcerative colitis, primary sclerosing cholangitis, idiopathic thrombocytopenic purpura, Sjögren’s syndrome, sarcoidosis, antiphospholipid antibody syndrome, and systemic lupus erythematosus(10,11). The possible mechanism of association between these autoimmune diseases remains under intense scientific investigation(12).
Scleroderma is one of the diseases under investigation in this scenario. It is an immune-mediated disorder of connective tissues with a wide spectrum of clinical presentations, characterized by sclerosis resulting from collagen deposition(13). It can only affect skin, fascia, muscle, and bones, and in such cases it is characterized as localized scleroderma (LS), also known as morphea; alternatively, it is defined as systemic sclerosis (SS) when it expands and affects internal organs(14).
Of the several pathophysiological mechanisms proposed to explain the disease, the most accepted mechanism is as follows: circulating dysfunctional endothelial progenitor cells would exacerbate vascular deterioration, thereby generating vascular and SS(15).
Recent studies have demonstrated that dysregulation in the expression of aquaporins in dermal fibroblasts and in the microvascular endothelium may also be responsible for the fibrosis observed in these conditions. The aquaporin 1 channel, for example, participates in angiogenesis and in extracellular matrix remodeling, and it is highly expressed in patients with SS(16).
Another pathophysiological situation occurs with aquaporin 3 channels, which participate in skin healing and dehydration processes that are directly related to the pathophysiology of SS, leading to an underexpression of these channels being observed in these patients(17).
Furthermore, neurological involvement in patients with SS or LS was previously considered rare and incidental; however, new studies have shown that approximately 40% of these patients present this association, ranging from mild headaches to severe myopathies. The inflammatory activity of SS is theorized to contribute to the CNS damage observed in NMOSD18.
It is evident that these diseases have a complex pathophysiology that is known to be associated with immunological and inflammatory mechanisms17, and share receptors from the same family, with similar structure and functions, in addition to still unknown factors. Therefore, the purpose of this study was to assess whether-in a manner similar to other autoimmune and immune-mediated diseases-systemic and localized sclerosis would be diseases with a significant correlation with NMO/ NMOSD, to open new study perspectives for these autoimmune diseases.
METHODS
This is an integrative literature review. The articles were selected from the MEDLINE (Online System of Search and Analysis of Medical Literature), SciELO (Scientific Electronic Library Online), LILACS (Latin American and Caribbean Literature in Health Sciences), and Scopus databases. All articles published in Portuguese, English, or Spanish in the last 20 years are selected. The keywords used were “neuromyelitis optica,” “systemic scleroderma,” and “localized scleroderma,” as well as their respective synonyms and terms in Portuguese and Spanish. Clinical studies, case reports, cohort studies, and case-control studies were included. Integrative reviews, narratives, letters to the editor, and articles unavailable in full text were excluded.
RESULTS
The search generated a total of 23 articles (7 articles in the Pan American Health Organization’s Virtual Health Library [VHL] search engine, which provides access to MEDLINE, LILACS, and SciELO, and 16 in the Scopus database). After the exclusion of duplicate articles, the articles obtained were read, eliminating those that did not match the subject or did not meet the established inclusion criteria (Figure 1).
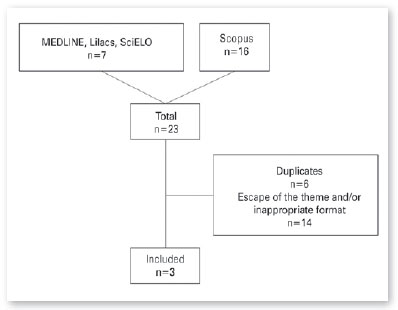
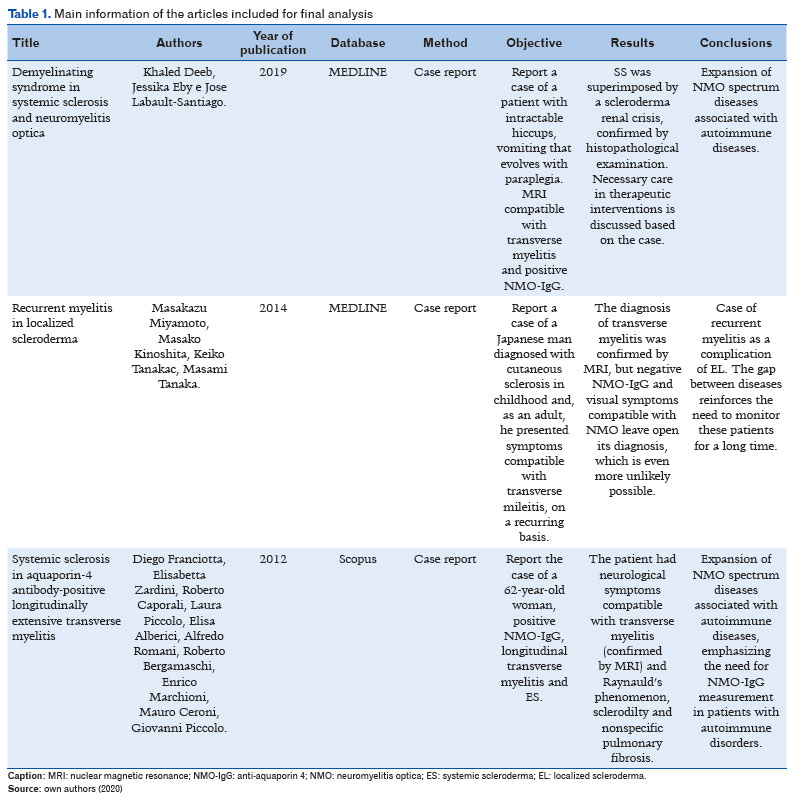
Finally, 3 articles were selected, whose main data will be summarized in Table 1 below.
DISCUSSION
Approximately 20%–30% of NMO carriers have other autoimmune disorders(20), of which systemic lupus erythematosus, Sjögren’s syndrome, rheumatoid arthritis, myasthenia gravis, and ulcerative colitis stand out as those with the highest correlation(1), but the association of NMO with SS and LS is somewhat infrequent. The present review focused its efforts on elucidating this gap.
Three case reports constitute this review’s final study material. The first case was a 44-year-old woman whose main complaint was the onset of intractable hiccups, vomiting, and dysphagia. The patient’s skin appeared to be thickened bilaterally at the metacarpophalangeal junctions, and she also presented Raynaud’s phenomenon. Subsequently, the patient developed blurred vision, muscle weakness, hypertonia, paroxysmal tonic spasms, Babinski’s sign, and paresthesia in the lower limbs. Magnetic resonance imaging (MRI) demonstrated hyperintense foci in the periventricular white matter and in the spinal cord between C2 and C6. Antinuclear antibodies, anti-RNA polymerase III, antitopoisomerase I, and NMO–IgG all tested positive, thereby establishing the diagnosis of NMO. Intravenous administration of methylprednisolone was initiated, which brought about a significant degree of clinical improvement. On the fifth day of hospitalization, the patient had a hypertensive crisis and acute kidney injury, which required urgent hemodialysis. A biopsy was performed, confirming the diagnosis of a corticosteroid-induced scleroderma renal crisis. The patient was subsequently treated with other drugs, responding well to rituximab(18).
Another case was a 39-year-old woman with a history of NMOSD since 12 years before the study duration, when she had quadriplegia, MRI with cervical transverse myelitis, and positive anti-AQP4. On that occasion, she responded positively to pulse therapy with methylprednisolone and azathioprine. She was then referred for rheumatologic evaluation owing to skin thickening and Raynaud’s phenomenon 1 year prior. Physical examination revealed telangiectasias on the face, leukomelanoderma on the chest, digital microulcerations, and diffuse skin thickening, with a modified Rodnan skin score of 22(17).
The patient had antinucleolar factor >1/1280, negative anti-Scl70, and a scleroderma pattern in nailfold capillaroscopy. Visceral involvement was ruled out, and the diagnosis of diffuse SS was confirmed using the ACR/EULAR criteria (2013). Monthly intravenous cyclophosphamide was then introduced, and after 8 months of treatment there was significant degree of skin improvement(17).
Finally, the last case analyzed was the report of a HCCP 54-year-old man, smoker, hypertensive, who had undergone a prostatectomy for cancer 2 years prior. Nearly 8 months previously, the patient presented rapidly progressing plaque scleroderma in the lower and upper limbs and in the abdomen. A skin biopsy was compatible with LS. Six months after the initial evaluation, the patient presented lower-limb paraparesis, constipation, and urinary retention, sensory level on T3, grade 0 strength in the lower limbs, and positive Babinski sign in bilateral extension. No eye complaints were reported. MRI showed a demyelinating lesion in the spinal cord, and anti-NMO was positive, confirming the diagnosis of NMOSD. The patient underwent pulse therapy with 1 g methylprednisolone for 5 days, 1 g cyclophosphamide, 2g/kg immunoglobulin, and plasmapheresis. After 14 days of hospitalization, a slight improvement in symptoms was observed(19).
The small number of viable articles selected for the final analysis of this review, and the fact that all were case reports is an indicator of the rarity of research on the association between NMO, SS, and LS. Other studies confirm this rarity, such as a Brazilian study performed with 22 patients with NMO, in which only one patient presented Raynaud’s phenomenon, with no description of SS or LS. The positivity of NMO–IgG antibodies within the sample was the most significant finding, as expected. However, antinucleosome, antinuclear, antithyroperoxidase, and antithyroglobulin antibodies were limited to a small number of patients(23). In an extensive literature review that retrieved articles from 1976 to 2017 dealing with NMO-associated autoimmune diseases, only three cases of SS/LS were found(24).
Concluding the present integrative review, it is understood that NMO–IgG is present in many different autoimmune and immune-mediated diseases. However, the association between NMO, SS, and LS is seldom described in the available medical literature researched, reaching the level of a rarity. This can be explained by the infrequent NMO-IgG testing of patients with other autoimmune diseases, as well as the also infrequent investigation of other autoimmune diseases in patients with NMO(23). Furthermore, as observed in the reported cases, sometimes the manifestations of these diseases can occur years apart(19,24), and even if they are suspected, not all the necessary criteria for their diagnosis may be present. This may be attributable to the use of immunosuppressive medications(20), which is common in these patients, or it may be because of an early stage of NMO, which may continue to be without seropositivity or exuberant clinical manifestations(24).
In view of the results reported in the articles included in this integrative review, it is understood that it is important to intensify efforts to conduct further research with designs that are capable of capturing more data in addition to those analyzed in this review. It would also be prudent and timely to investigate the association of NMO with other autoimmune and immune-mediated diseases, given the vast field of study and research available.
REFERENCES
1. Freitas E, Guimarães J. Neuromyelitis optica spectrum disorders associated with other autoimmune diseases. Rheumatol Int. 2015;35(2):243-53.
2. Sellner J, Boggild M, Clanet M, Hintzen RQ, Illes Z, Montalban X, et al. EFNS guidelines on diagnosis and management of neuromyelitis optica. Eur J Neurol. 2010;17(8):1019-32.
3. Fazio R, Radaelli M, Furlan R. Neuromyelitis optica: concepts in evolution. J Neuroimmunol. 2011;231(1-2):100-4.
4. Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet. 2004;364(9451):2106-12.
5. Jacob A, McKeon A, Nakashima I, Sato DK, Elsone L, Fujihara K, et al. Current concept of neuromyelitis optica (NMO) and NMO spectrum disorders. J Neurol Neurosurg Psychiatry. 2013; 84(8):922-30.
6. González C, González-Buitrago JM, Izquierdo G. Aquaporins, anti-aquaporin-4 autoantibodies and neuromyelitis optica. Clin Chim Acta. 2013;415:350-60.
7. Misu T, Höftberger R, Fujihara K, Wimmer I, Takai Y, Nishiyama S, et al. Presence of six different lesion types suggests diverse mechanisms of tissue injury in neuromyelitis optica. Acta Neuropathol. 2013;125(6):815-27.
8. Ingegnoli F, Gualtierotti R, Pierro L, Del Turco C, Miserocchi E, Schioppo T, et al. Choroidal impairment and macular thinning in patients with systemic sclerosis: the acute study. Microvasc Res. 2015;97:31-6.
9. Pittock SJ, Lennon VA, De Seze J, Vermersch P, Homburger HA, Wingerchuk DM, et al. Neuromyelitis optica and non-organ-specific autoimmunity. Arch Neurol. 2008;65(1):78-83.
10. Jarius S, Jacobi C, De Seze J, Zephir H, Paul F, Franciotta D, et al. Frequency and syndrome specificity of antibodies to aquaporin-4 in neurological patients with rheumatic disorders. Mult Scler J. 2011;17(9):1067-73.
11. Kolfenbach JR, Horner BJ, Ferucci ED, West SG. Neuromyelitis optica spectrum disorder in patients with connective tissue disease and myelitis. Arthritis Care Res (Hoboken). 2011;63(8): 1203-8.
12. Wingerchuk DM, Weinshenker BG. The emerging relationship between neuromyelitis optica and systemic rheumatologic autoimmune disease. Mult Scler J. 2012;18(1):5-10.
13. Zulian F, Cuffaro G, Sperotto F. Scleroderma in children: an update. Curr Opin Rheumatol. 2013;25(5):643-50.
14. Arango C, Malagón C, Gómez M del P, Mosquera C, Yépez R, González T, et al. Esclerodermia localizada juvenil:¿ es una enfermedad benigna? Rev Colomb Reumatol. 2017;24(3):145-52.
15. Trojanowska M. Cellular and molecular aspects of vascular dysfunction in systemic sclerosis. Nat Rev Rheumatol. 2010; 6(8):453.
16. Yamashita T, Asano Y, Saigusa R, Taniguchi T, Nakamura K, Miura S, et al. Increased expression of aquaporin-1 in dermal fibroblasts and dermal microvascular endothelial cells possibly contributes to skin fibrosis and edema in patients with systemic sclerosis. J Dermatol Sci. 2019;93(1):24-32.
17. Farhadi E, Mahmoudi M, Rahmani F, Yousefi B, Sarafnejad A, Kavosi H, et al. Attenuation of aquaporin-3 and epidermal growth factor receptor expression and activation in systemic sclerosis dermal fibroblasts. J Cell Physiol. 2019;234(8):12876-83.
18. Amaral TN, Peres FA, Lapa AT, Marques-Neto JF, Appenzeller S. Neurologic involvement in scleroderma: a systematic review. In: Seminars in arthritis and rheumatism. Elsevier; 2013. p. 335-47.
19. Teixeira JMA, Oliveira JL, Cordeiro RA, Luppino-Assad AP, Sampaio-Barros PD. Doença do espectro da neuromielite óptica e esclerose sistêmica: uma rara associação. Rev Bras Reumatol. 2017;57:S89-90.
20. Iyer A, Elsone L, Appleton R, Jacob A. A review of the current literature and a guide to the early diagnosis of autoimmune disorders associated with neuromyelitis optica. Autoimmunity. 2014;47(3):154-61.
21. Deeb K, Eby J, Labault-Santiago J. Demyelinating syndrome in systemic sclerosis and neuromyelitis optica. BMC Neurol. 2019; 19(1):1-6.
22. Maciel ML, Sa AT, Zimmermann AF, Neves FS, Fialho S, Pereira IA. Doença do espectro da neuromielite óptica com anti-aquaporina-4 associado a morfeia (esclerodermia localizada). Rev Bras Reumatol. 2017;57:S264-5.
23. Pereira WLDCJ, Reiche EMV, Kallaur AP, Oliveira SR, Simão ANC, Lozovoy MAB, et al. Frequency of autoimmune disorders and autoantibodies in patients with neuromyelitis optica. Acta Neuropsychiatr. 2017;29(3):170-8.
24. Shahmohammadi S, Doosti R, Shahmohammadi A, Mohammadianinejad SE, Sahraian MA, Azimi AR, et al. Autoimmune diseases associated with neuromyelitis optica spectrum disorders: a literature review. Mult Scler Relat Disord. 2019;27:350-63.
AUTHOR’S INFORMATION







How to cite: No specific financial support was available for this study
Disclosure of potential conflicts of interest: None of the authors have any potential conflict of interest to disclose
Received on:
June 24, 2021.
Accepted on:
May 24, 2022.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados
 Ler em português
Ler em português
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket