

Caroline Oliveira Brêtas; Thiago George Cabral Silva; Ledilma Inês Colodetti Zanandrea; Patrícia Grativol Costa Saraiva; Fábio Petersen Saraiva
DOI: 10.17545/eOftalmo/2020.0002
RESUMO
Vasculites retinianas representam um verdadeiro desafio na prática oftalmológica. O diagnóstico precoce e tratamento adequados são de extrema importância para obter bom prognóstico visual e evitar complicações. O objetivo deste artigo é elaborar um protocolo para investigação de vasculites retinianas para facilitar o diagnóstico e conduta.
Palavras-chave: Vasculite retiniana; Uveíte; Protocolos clínicos.
ABSTRACT
Retinal vasculitis represents a real challenge in ophthalmic practice. Eartly diagnosis and treatment are important for good visual prognosis and to avoid complications. The aim of this article is to develop a protocol for investigation of retinal vasculitis to facilitate diagnosis and management.
Keywords: Retinal vasculitis; Uveitis; Clinical protocols.
INTRODUÇÃO
Vasculites retinianas são alterações inflamatórias dos vasos retinianos que podem estar associadas à doença ocular primária ou à doença sistêmica, seja ela idiopática, inflamatória, imunomediada, infecciosa ou maligna. A patogênese da vasculite retiniana é presumivelmente um fenômeno autoimune, com evidências da presença de linfócitos TCD4+ dentro ou entorno dos vasos sanguíneos(1). A incidência é estimada em 1-2 novos casos a cada 100.000 habitantes nos Estados Unidos(2). A forma bilateral é a mais comum(3).
A vasculite pode se apresentar como hemorragia intraretiniana, mancha algodonosa, embainhamento vascular, oclusão vascular ou vazamento de líquido(4). Além disso, pode existir ou não predominância arteriolar ou venular, sendo este um dado importante durante a investigação etiológica(5). Podem complicar com edema macular, neovascularização, hemorragia vítrea, descolamento de retina, membrana epirretiniana e glaucoma neovascular(5) (Figura 12).
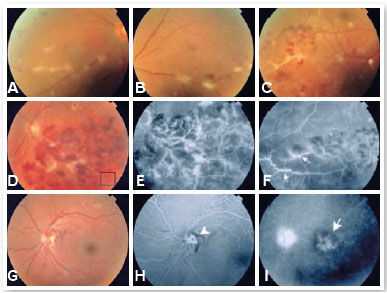
Cerca de 14,9% dos pacientes com uveíte apresentam evidências de vasculite retiniana em suas diversas etiologias segundo Rosenbauma et al.(6). A associação com doença de Behçet é comum, enquanto espondilite anquilosante e artrite idiopática juvenil são raras(6). No caso de doenças reumáticas, é importante a avaliação oftalmológica tendo em vista que as vasculites retinianas podem ser assintomáticas principalmente se de início precoce ou periféricas(7). Além disso, podem ser a primeira manifestação de uma doença sistêmica. O diagnóstico precoce é chave para o sucesso do tratamento e prognóstico(7).
MÉTODOS
Este artigo apresenta uma revisão da literatura não sistemática. Para levantamento bibliográfico realizou-se busca de artigos nas bases de dados: Pubmed e Scielo. Os descritores utilizados foram: Vasculite Retiniana; Uveíte; Protocolos Clínicos.
REVISÃO DA LITERATURA
Epidemiologia e classificação
Em relação à etiologia podem ser: idiopáticas, representar uma doença ocular primária ou estar associada a alguma doença sistêmica, infecção, síndrome oclusiva ou ainda tumores (síndrome mascarada)(1).
Vasculite retiniana idiopática, aneurismas e neurorretinite (IRVAN)
Descrita em 1995 por Chang et al, é rara e de etiologia desconhecida. Afeta mais comumente pacientes jovens, hígidos e mulheres. Não há predileção racial.
Alguns autores descreveram associação com vasculites sistêmicas que cursam com anticorpo anti-citoplasma de neutrófilos perinuclear (p-ANCA)(8,9).
O diagnóstico contempla três critérios maiores (vasculite retiniana, dilatações aneurismáticas nas bifurcações arteriais e neurorretinite) e três menores (não perfusão capilar periférica, neovascularização e exsudação macular)(8). Os pacientes apresentam baixa acuidade visual em um ou ambos os olhos e vitreíte de intensidade variável. As arteríolas apresentam aneurismas que se estendem das primeiras bifurcações até média periferia; comumente encontrados no disco óptico. A exsudação lipídica é acentuada e tipicamente encontrada na região peripapilar podendo se estender até a mácula, cursando com estrela macular(10).
Doenças oculares primárias
Entre as doenças oculares primárias destacam-se doença de Eales, pars planitis ou uveíte intermediária e retinocoroidopatia de Birdshot. Menos comumente podemos citar Síndrome de Vogt-Koyonagi-Harada (SVKH) e Oftalmia Simpática(1).
A doença de Eales é uma vasculopatia oclusiva retiniana idiopática que acomete bilateralmente indivíduos jovens de 20-45 anos e saudáveis. Observa-se periflebite e não perfusão capilar retiniana periférica, preferencialmente no quadrante temporal superior. Sua etiologia é desconhecida e já foi relatada associação com hipersensibilidade à proteína tuberculínica. Os pacientes que apresentam neovascularização periférica sem nenhuma outra causa específica são designados de “doença de Eales”(11). Pode complicar com hemorragia vítrea, descolamento de retina tracional ou regmatogênico, rubeosis iridis, glaucoma secundário e catarata(11). O tratamento inclui fotocoagulação da retina isquêmica e uso de corticosteroides sistêmico ou ocular. O prognóstico é bom em geral(11).
A retinocoroidopatia de Birdshot é uma uveíte posterior crônica bilateral caracterizada por difusa inflamação da coroide associada à vitreíte e vasculite retiniana. É rara, de causa desconhecida e associada ao HLA-A29. É mais comum em mulheres caucasianas entre 3ª e 6ª décadas. Acomete o estroma da coroide, com lesões despigmentadas da cor creme e de formato oval, com margens mal definidas e em diferentes estágios de evolução localizados no pólo posterior e média periferia. Apresenta-se com reação de câmara anterior mínima, vitreíte, extravasamento vascular com edema macular e às vezes edema de disco óptico(1). As lesões quando ativas tem consistência e, ao cicatrizarem, deixam manchas hipopigmentadas ou desaparecem. O tratamento inclui: corticosteroides e imunossupressores(1).
Doenças sistêmicas
Entre as doenças sistêmicas comumente relacionadas com vasculites retinianas, merecem destaque: doença de Behçet, sarcoidose, lúpus eritematoso sistêmico e Granulomatose de Wegener. Doenças como esclerose múltipla, poliarterite nodosa, doenças inflamatórias intestinais e outras doenças reumáticas também podem estar associadas(1).
A doença de Behçet é a doença reumática mais associada à vasculite retiniana5. É uma doença multissistêmica inflamatória com associação com HLA-B51. Apresenta episódios recorrentes de vasculite oclusiva acometendo principalmente veias de pequeno calibre e se manifestando com úlceras orais e genitais, lesões cutâneas e inflamação ocular. A doença é mais grave em homens. Manifestações oculares estão presentes em 50-85% dos casos, bilaterais em 75-94%, sendo o olho o principal órgão acometido na doença. A vasculite retiniana pode-se apresentar com embainhamento vascular e oclusões vasculares, edema, exsudatos, hemorragias e infiltrados frequentemente associados à vitreíte. Pode haver iridociclite e hipópio(12).
A sarcoidose é uma doença inflamatória multissistêmica crônica de etiologia desconhecida caracterizada histologicamente pela formação de granulomas não caseosos. A inflamação ocular é geralmente bilateral e muitas vezes granulomatosa. Acomete adultos jovens de 20-40 anos. A doença sistêmica predomina em mulheres, no entanto o acometimento ocular não apresenta predileção quanto ao sexo. É predominantemente uma periflebite e com descontinuidade. As alterações do segmento posterior incluem: uveíte intermediária com vitreíte, snowballs e/ou snowbanks, vasculite periférica (embainhamento perivenular, exsudatos perivensos em cera de vela candle wax drippings), granulomas coroidianos ou do disco óptico, nódulos pré-retinianos (Sinal de Landers) e pequenas cicatrizes atróficas numulares na periferia retiniana(13).
A poliarterite nodosa é uma doença incomum caracterizada por vasculite necrotizante de artérias de pequeno e médio calibre com particular predileção por artérias renal e visceral. Sua importância consiste no fato de poder comprometer as artérias ciliares posteriores e vasos da coroide e a vasculite acarretar infarto da coroide e descolamento de retina secundário(1).
A poliangeíte microscópica (PAM) é uma vasculite necrotizante que acomete a microvascultura (arteríolas, capilares e vênulas) e raramente médias artérias. É rara e autoimune, associada à presença do anticorpo anticitoplasma de neutrófilos (ANCA), com predomínio do padrão p-ANCA sobre o c-ANCA. Existe relato de associação com oclusão de artéria central da retina(14).
O acometimento ocular no lúpus eritematoso sistêmico (LES) pode refletir a atividade sistêmica da doença e a gravidade, além de poder ser o primeiro sintoma relatado. A prevalência da retinopatia lúpica varia de 3% em pacientes ambulatoriais com doença leve a ausente para 29% em pacientes com doença em atividade. Além disso, a presença de anticorpos antifosfolípideos está associada ao aumento na prevalência de retinopatia vaso-oclusiva e oclusões vasculares, principalmente de artéria central da retina. Apesar de a preferência ser por artérias, existem relatos de oclusão de veia central da retina associada(7,15).
O envolvimento retiniano na Granulomatose de Wegener (GW) é incomum (5-12%), apresentando-se mais comumente como hemorragias na periferia da retina. Está associada à anormalidade arterial e venosa, hemorragias, edema e manchas algodonosas, podendo também acometer nervo óptico e cursar com edema de disco(1,7).
Em pacientes com artrite reumatoide foi relatada uma prevalência de 16% de manifestações oftalmológicas. As mais comuns são esclerite, esclerite necrotizante e PUK. As duas últimas indicam a presença de vasculite sistêmica em atividade e aumento de mortalidade. A vasculite retiniana e coroidite são eventos raros(7).
No caso das espondiloartropatias, o acometimento do seguimento anterior do olho é mais comum e o posterior pode ocorrer em até 20% dos pacientes. A presença do HLAB27 em paciente com uveíte confere maior risco de complicações em comparação com grupo de uveíte anterior idiopática(7,16).
A doença de Crohn é uma doença inflamatória transmural granulomatosa do intestino caracterizada por preferencialmente íleo distal. É a doença inflamatória intestinal mais comum associada a manifestações extraintestinais. Cerca de 5-10% apresentam complicações oculares, incluindo ceratite, episclerite, iridociclite e edema macular. Existem relatos de associação com oclusão de artéria central da retina e vasculite de nervo óptico(17).
Doenças infecciosas
As causas infecciosas podem ser dividas em bacterianas, como sífilis e tuberculose, virais como o herpes, vírus da imunodeficiência humana e citomegalovírus e parasitárias como a toxocaríase e toxoplasmose. Nos casos de pacientes imunossuprimidos com vasculite sistêmica que apresentam vasculite retiniana, a causa mais comum é a infecciosa em detrimento da doença de base(5).
A sífilis ocular pode-se manifestar de diversas formas tendo por isso recebido a referência “grande imitadora”. A principal forma é a panuveíte, podendo também ser: ceratite, irite, neurite óptica, edema macular, vitreíte e retinite. Uma apresentação rara e grave é a oclusão de artéria central(18).
No caso das vasculites relacionadas à síndrome mascarada, lembrar-se de linfoma não Hodgkin do sistema nervoso central, leucemia, melanoma, metástase intraocular de carcinomas, retinoblastoma e embolização sistêmica de placas de colesterol(19,20).
Segue abaixo os principais diagnósticos de acordo com a classificação etiológica(1):
Classificação etiológica
• Idiopática
– Síndrome de IRVAN
• Doenças oculares primárias
– Doença de Eales
– Retinocoroidopatia Birdshot
– Uveíte intermediária
– Síndrome de Vogt-Koyanagi-Harada
– Oftalmia Simpática
– Vasculite retiniana multifocal aguda
• Doenças sistêmicas
– Doença de Behçet
– Sarcoidose
– Esclerose múltipla
– Granulomatose de Wegener
– Lúpus eritematoso sistêmico
– Poliarterite nodosa
– Poliangeite microscópica
– Doença de Crohn
– Espondilite anquilosante
– Doença de Buerger
– Policondrite recidivante
– Síndrome antifosfolípide
– Síndrome de Churg-Strauss
– Polimiosite
– Artrite Reumatóide
– Dermatomiosite
– Doença de Takayasu
• Causas infecciosas
– Bacterianas: sífilis, tuberculose, doença de Lyme, doença de Whipple, brucelose, doença da arranhadura do gato, ricketssiose
– Virais: herpes simples, varicela zoster, citomegalovírus, HIV, HTLV, hepatites, vírus Epstein-Barr, vírus da dengue
– Parasitárias: toxocaríase e toxoplasmose
– Endoftalmite fúngica
• Doenças oclusivas
– Trombofilias
– Hemoglobinopatias
• Síndrome mascarada
– Tumores: linfoma primário de SNC, linfoma vítreo-retiniano, leucemia aguda, retinopatia associada ao câncer (CAR)
– Metástases
MANIFESTAÇÕES CLÍNICAS
A vasculite pode-se apresentar assintomática em um primeiro momento, especialmente quando as alterações vasculares se desenvolvem na periferia da retina(5).
Os sintomas clínicos incluem: turvação visual indolor, alteração na percepção de cores, metamorfopsias, floaters (moscas volantes) e escotomas. Menos comumente pode haver diminuição da visão de cores e dor. Sinais e sintomas de envolvimento sistêmico podem estar presentes merecendo destaque: úlceras orais, genitais e de pele, artrite, rash, doença neurológica e evidência de doença embólica(1,5,7).
O embainhamento vascular é a característica mais comum presente na biomicroscopia de fundo, além de manchas algodonosas, vitreíte de intensidade variável, snowballs, descolamento do vítreo, hemorragias, edema macular, neovascularização e quadro de uveíte anterior. É importante destacar que nem todos os casos de embainhamento vascular são causados por vasculite e o embainhamento vascular congênito é um exemplo. É de ocorrência comum e normalmente presente dentro dos dois diâmetros de disco do nervo óptico, associado mais a veia e pode estar presente com persistência da vasculatura hialoidea. O haze vítreo traduz a quebra da barreira hematorretiniana, porém inespecífica de processo inflamatório; já a celularidade vítrea é sinal de condição inflamatória(1).
Inflamação coroideana sugere quadro de sarcoidose, retinocoroidopatia de Birdshot ou síndrome de histoplasmose ocular(1).
As alterações tardias secundárias à oclusão e remodelamento incluem: telangectasias, microaneurismas, oclusão de ramo venoso ou veia central, neovascularização isquemia-induzida; podendo cursar com sequelas, como hemorragia vítrea, descolamento tracional de retina, rubeosis de íris e glaucoma neovascular(1).
De acordo com as manifestações clínicas, pode-se também classificar as vasculites retinianas em(1):
• Infecciosa ou não infecciosa
• Arterial, venosa ou mista
• Unilateral ou bilateral
• Pólo posterior ou periferia de retina
• Focal segmentar ou difusa
• Oclusiva ou não oclusiva
Anamese
A anamnese deve ser criteriosa com intuito de identificar fatores de risco e antecedentes patológicos pessoais ou familiares que possam direcionar quanto à etiologia.
A seguir destacamos, em tópicos, dados importantes a serem coletados na anamnese, correlacionando-os com a possível etiologia(1,21).
• Claudicação de mandíbula, hipersensibilidade no couro cabeludo, polimialgia reumática, artéria temporal palpável
– Arterite de células gigantes (Takayasu)
• Perdas gestacionais de repetição, tromboses recorrentes
– Trombofilias
– Sindrome do anticorpo fosfolípide
– Hemoglobinopatias
• Sintomas articulares
– Espondilite Anquilosante
– LES
– Poliarterite nodosa
– Policondrite recidivante
– Buerger
– Artrite Reumatoide
• Sintomas neurológicos
– Esclerose múltipla
• Sintomas intestinais
– Doença de Crohn ou retocolite ulcerativa
• Úlceras orais e genitais, teste da patergia
– Doença de Behçet
• História ou suspeita de neoplasia maligna
– Síndrome mascarada
• Transfusões sanguíneas prévias
– Hepatites B e C
– HIV
– HTLV
Além desses dados, é importante investigar história de tabagismo e uso contraceptivos orais que podem estar associados a síndromes oclusivas.
Exame oftalmológico
O padrão de envolvimento vascular e sua localização podem auxiliar no diagnóstico, etiologias diferentes geralmente manifestam tropismo para diferentes tipos de vaso sanguíneo, a exemplo podemos citar o LES e GW que acometem artérias e Behçet, sarcoidose e esclerose múltipla com preferência em veias(5). Os principais diagnósticos de acordo com o padrão vascular acometido podem ser encontrados na tabela 1(1).
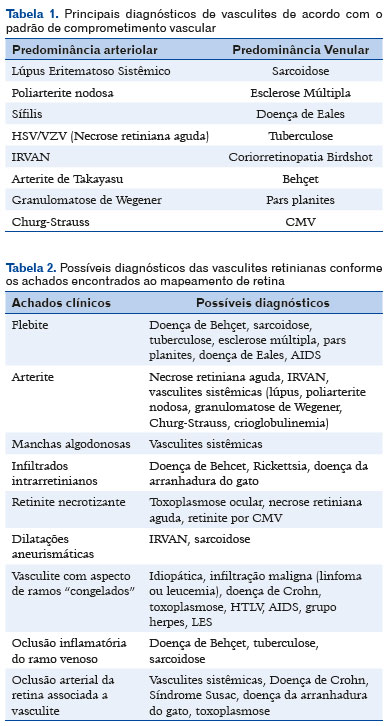
Outras alterações oftalmológicas observadas ao mapeamento de retina também podem auxiliar no direcionamento da diagnose da causa da vasculite retiniana (Tabela 2)(1).

Exames complementares
A vasculite pode ser detectada clinicamente ou por um exame complementar como a angiografia fluoresceínica (AF) em casos com sinais mínimos ou subclínicos(7). A AF evidencia presença de corante estagnado no vaso ou extravasamento deste através da parede vascular(1,5). O vazamento do corante leva à hiperfluorescência progressiva da parede do vaso na doença em atividade e evidencia o aumento da permeabilidade vascular. No caso de vasculite do nervo óptico, observa-se edema na cabeça do disco e neuropatia óptica isquêmica(7). As manchas algodonosas são vista na AF como áreas de não perfusão focais. A tomografia de coerência óptica (OCT) é útil para detectar e confirmar a presença de fluido subrretiniano nos casos que evoluem com edema macular(1). A biópsia é raramente é realizada, tendo em vista o dano potencial que causa na retina(1,5). Exames laboratoriais são fundamentais para o diagnóstico etiológico. Portanto, diante de um caso novo, considerando as causas mais comuns de vasculites retinianas e na ausência de uma doença de base já definida, recomenda-se solicitar:
• Retinografia
• Tomografia de Coerência Óptica (OCT)
• Angiografia com fluoresceína
• Exames gerais
– Aferição da pressão arterial
– Hemograma
– Plaquetas
– Glicemia de jejum
– Ureia e creatinina
– EAS
• Sorologias para doenças infecciosas
– HIV 1 e 2
– HTLV
– VDRL
– FTAbs
– Hepatite A, B e C
– Toxoplasmose IgM IgG
– CMV IgM IgG
– Herpes IgM IgG
– Toxocaríase IgM IgG
– Teste tuberculínico (PPD)
– Bartonella henselea
• Sorologias para doenças auto-imunes
– VHS, PCR
– FAN, c-ANCA, p-ANCA, Anti-Sm, Anti-dsDNA
– ASCA
– FR, anti-CCP
– HLA (HLA-B27/ HLA-A29/ HLA-B51)
– Anticoagulante lúpico, Anticardiolipina IgM/IgG, Antifosfolípide
– C3, C4, CH50
– Enzima Conversora de Angiotensina (ECA), lisozima
Na ausência de confirmação diagnóstica com tais exames ou dependendo de dados da anamnese, pode-se ainda complementar a investigação com:
• Colonoscopia e Endoscopia
– História de sintomas intestinais (doença de Crohn e Retocolite ulcerativa)(17)
• RNM de encéfalo (e, se necessário coluna cervical, torácica e lombar) e análise de liquor(1)
– História de déficit neurológico, aumento de IgG no líquor (Esclerose múltipla)(1)
• Biópsia de artéria temporal(20)
– Claudicação de mandíbula, polimialgia reumática, artéria temporal palpável, hipersensibilidade do couro cabeludo (Arterite Temporal)
• Radiografia de tórax e Tomografia computadorizada de tórax
– Linfadenopatia hilar bilateral, paratraqueal ou infiltrados pulmonares parenquimatosos, fibrose (Sarcoidose)(1,13)
• Linfadenomegalia mediastinal, caverna primária, tuberculomas calcificados e infiltrados pulmonares, micronódulos difusos (Tuberculose)(1,22)
– Tumores sistêmicos, locais e implantes secundários (Síndrome Mascarada)(19)
• Arteriografia, Angiotomografia ou Angioressonância
– Aneurismas e microaneurismas em artérias médias, predileção para artéria renal e visceral (Poliarterite nodosa)(23)
• Ultrassonografia ocular
– Massas intraoculares (Tumores)(1,19)
• Biópsias de vasos(1,21)
• Eletroforese de Hb(21)
– Hemoglobinopatias
• Dosagem de bilirrubinas(1)
– Alteração hepática, hemólise (anemia, hepatite)
• Coagulograma (fatores VII, IX, XI); Proteína C e S; Fator V de Leiden; Mutação do gene da protrombina; Homocisteína(1)
• Investigação de trombofilia(1)
• Sorologias para dengue, rickettsias, doença de Lyme, doença da arranhadura do gato(1)
• PCR do humor aquoso(1)
– Biópsia vítrea (casos com suspeita de malignidade)(1,19)
Na presença de uma sorologia infecciosa específica, pode-se fechar um diagnóstico de presunção. Testes imunológicos específicos usualmente estão positivos em apenas determinadas doenças. As doenças abaixo relacionadas estão associadas a positividades dos seguintes exames:
• LES: FAN, Anti-Sm, Anti-dsDNA, CH50, HLA-DR3, C3 e C4 diminuídos(13,15,21)
• Síndrome do anticorpo antifosfolípide: anticoagulante lúpico, anticardiolipina, antifosfosfolípide(15)
• Behçet: Teste da patergia, HLA-B51(12)
• Poliarterite nodosa: sorologia de hepatites, (15-30% dos pacientes tem infecção pelo vírus da hepatite B; 5% da hepatite C), c-ANCA, p-ANCA(23,24)
• Sarcoidose: enzima conversora de angiotensina (ECA), lisozima, aumento de cálcio sérico(13)
• Artrite Reumatoide: FR, anti-CCP(21)
• Espondilite Anquilosante: HLA-B27(7)
• Birdshot: HLA-A29(21)
• Granulomatose de Wegener: c-ANCA, p-ANCA(24)
• Síndrome de Churg-Strauss: c-ANCA, p-ANCA(23,24)
• Doença de Crohn: ASCA(25)
• Retocolite Ulcerativa: p-ANCA(25)
Apresentamos em anexo um fluxograma (A) como proposta para condução de um caso novo de vasculite retiniana.
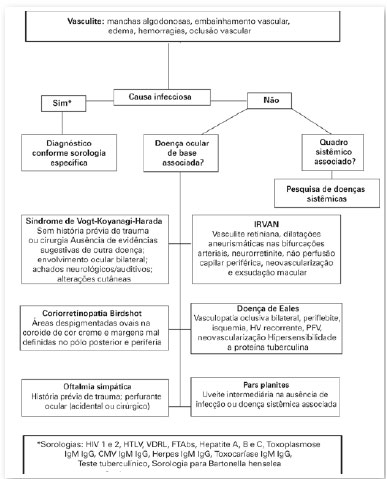
TRATAMENTO
O tratamento é direcionado para a causa, sinais e sintomas do paciente. O objetivo é suprimir a inflamação intraocular para prevenir perda visual e complicações em longo prazo, além de manter a remissão e evitar recaídas. A terapia pode não ser requerida em casos com atividade mínima de doença e acuidade visual de 20/40 ou melhor. Em casos de inflamação leve, boa acuidade visual e ausência de envolvimento retiniano extenso, pode-se optar pela observação ou uso de corticoterapia tópica ou subtenoniana.
Nos casos mais graves, com perda importante da acuidade visual, edema macular e acometimento retiniano extenso, o uso de corticoterapia sistêmica (oral ou venoso) está indicado. Uma vez obtido controle da doença, deve ser feita redução gradativa da medicação. Fármacos imunossupressores como a azatioprina, metotrexato e ciclosporina podem ser tentados em casos refratários. Vitrectomia e fotocoagulação a laser estão indicados em casos de neovascularização persistente, hemorragia vítrea ou glaucoma(1).
As causas infecciosas devem ser tratadas direcionadas ao agente etiológico. Em casos secundários a Doença de Behçet, opta-se por anticorpo monoclonal anti-TNF ou interferon-alfa. Vasculites oclusivas, a exemplo da Síndrome Anticorpo Antifosfolípidio, são tratadas com terapia anti-plaquetária e/ou anticoagulante.
Na maioria dos casos, o tratamento é realizado com os corticoesteroides locais ou sistêmicos em associação ou não a imunossupressores, e, posteriormente, terapia com imunobiológicos. Os corticoides são mais rapidamente eficazes; enquanto os imunossupressores podem demorar até 3 meses para fazer efeito(5).
A injeção de subtenoniana de corticoide é útil em pacientes com doença unilateral ou grave. A rota periocular evita os efeitos colaterais sistêmicos da via oral, apesar do risco de aumento da pressão intraocular, catarata ou perfuração ocular acidental. A melhora clínica é observada em 2 a 3 semanas após o procedimento e os efeitos podem durar até 3 meses.
A corticoterapia oral é tipicamente utilizada em casos moderados a graves bilaterais com redução importante da acuidade visual. A dose recomendada é 1-2mg/kg. Alguns autores recomendam o regime de alta dose por 3-4 semanas enquanto outros defendem a redução da dose em 50% a cada 8 dias seguido de 1 mês de dose reduzida. Caso não apresente recorrência o corticoide poderá ser mantido a longo prazo numa dose de 7,5mg/dia.
Os imunossupressores devem ser considerados em casos insuficientemente ou não responsivos e efeitos colaterais aos corticoides. Os agentes que podem ser utilizados são: azatioprina, ciclofosfamida, ciclosporina, metotrexato, clorambucil, micofelonato mofetil e tacrolimus. Destes, a azatioprina e ciclosporina são os mais empregados.
➞ Ciclosporina: 2,5 a 5mg/kg/dia dividida em 2 doses diárias.
➞ Azatioprina: 1 a 3mg/kg/dia
O uso de anticorpos anti-fator do crescimento do endotélio vascular (anti-VEFG), assim como corticoides na forma de injeção intravítrea, pode ser tentado para o manejo de complicações, como o edema macular.
O fluxograma de tratamento das vasculites segue em anexo (Fluxograma B).

PROGNÓSTICO
O prognóstico visual é variável. Alguns apresentam curso benigno e com boa resposta à terapia, preservando a acuidade visual. Porém, quadros mais resistentes mesmo com imunossupressores, podem evoluir com perda da função visual, tornando o prognóstico visual reservado.
REFERÊNCIAS
1. Siqueira RC, Oréfice F. Vasculites da Retina. In: Oréfice, F. et al. Uveítes. Série Oftalmologia Brasileira. 4ª edição, Rio de Janeiro, Cultura Médica, 2016. Cap.11, p. 105-123.
2. Hughes EH, Dick AD. The pathology and pathogenesis of retinal vasculitis. Neuropathol Appl Neurobiol. 2003;29(4):325-40.
3. Ali A, Ku JH, Suler EB, el al. The course of retinal vasculitis. Br J Ophthalmol. 2014;98(6):785-9.
4. Tahreem AM, et al. Clinical Features and incidence rates of ocular complications in patients with retinal vasculitis. Am J Ophthalmol. 2017;179:171-8.
5. Rosenbauma JT, Sibley CH, Lin P. Retinal Vasculitis. Curr Opin Rheumatol. 2016;28(3):228-35.
6. Rosenbauma JT, Ku J. Ali, A., et al. Patients with Retinal Vasculitis rarely suffer from systemic vasculitis. Semin Arthritis Rheum. 2012;41(6):859-65.
7. Androudi S, Dastiridou A, Symeonidis C, et al. Retinal vasculitis in rheumatic diseases: na unseen burden. Clin Rheumatol. 2013; 32(1):7-13.
8. Chang TS, Aylward GW, Davis JL, et al. Idiopathic retinal vasculitis, aneurysms, and neuro-retinitis: Retinal Vasculitis Study. Ophthalmology. 1995;102(7):1089-97.
9. Soheilian M, Nourinia R, Tavallali A, et al. Idiopathic retinal vasculitis, aneurysms, and neuroretinitis syndrome associated with positive perinuclear antineutrophil cytoplasmic antibody (P-ANCA). Retin Cases Brief Rep. 2010;4:198-201.
10. Agarwal A. Idiopathic retinal vasculitis, aneurysms, and neuroretinopathy (IRVAN). In: Gass’ Atlas of Macular Diseases. 5ª edição. Philadelphia: Saunders: 2012, p.534-538.
11. Jr Mathura JR, Bearelly S, Jampol LM. Retinopatias Proliferativas. In: Yanoff, M.; Ducker J.S.; Retina e Vítreo. 4ª edição, Rio de Janeiro, Elsevier, 2017. Cap. 26, p.152-156.
12. Oréfice F, Diligente FT, Oréfice JL. Doença de Behçet. In: Oréfice, F. et al. Uveítes. Série Oftalmologia Brasileira. 4ª edição, Rio de Janeiro, Cultura Médica, 2016. Cap.33, p. 369-377.
13. Oréfice F, Diligente FT, Cattan JM. Sarcoidose. In: Oréfice, F. et al. Uveítes. Série Oftalmologia Brasileira. 4ª edição, Rio de Janeiro, Cultura Médica, 2016. Cap.32, p. 361-367
14. Domingues CG, Capitanio CF, Abreu LB, et al. Central retinal artery occlusion in patient with microscopic polyangiitis. Rev Bras Oftalmol. 2015;74(6):386-9.
15. Seth G, Chengappa KG, Misra DP, et al. Lupus retinopathy: a marker of active systemic lupus erythematosus. Rheumatol Int. 2018;38(8):1495-501.
16. Power WJ, Rodriguez A, Pedroza-Seres M. Outcomes in anterior uveitis associated with the HLA-B27 haplotype. Ophthalmology. 1998;105(9):1646-51.
17. Coussa RG, Ali-Ridha A, Vila N. Simultaneous central retinal artery occlusion and optic nerve vasculitis in Crohn disease. Am J Ophthalmol Case Rep. 2017;5:11-5.
18. Khan MS, Kuruppu DK, Popli TA, et al. Unilateral optic neuritis and central retinal vasculitis due to ocular syphilis. Retin Cases Brief Rep. 2017
19. Oréfice F, Diligente FT, Cattan J M. Síndromes Mascaradas. In: Oréfice, F. et al. Uveítes. Série Oftalmologia Brasileira. 4ª edição, Rio de Janeiro, Cultura Médica, 2016. Cap.32, p. 425-438.
20. Jacobson DM. Systemic Cholesterol Microembolization Syndrome Masquerading as Giant Cell Arteritis. Survey of Ophthalmology. 1991;36(1):23-7.
21. Agarwal A, et al. Retinal Vasculitis. EyeWiki, São Francisco, CA, 16 de dez. 2019. Disponível em: <https://eyewiki.aao.org/Retinal_Vasculitis> Acesso em: 30 de fev de 2020.
22. Castro MC, Oréfice F. Tuberculose. In: Oréfice, F. et al. Uveítes. Série Oftalmologia Brasileira. 4ª edição, Rio de Janeiro, Cultura Médica, 2016. Cap.23, p. 233-253.
23. Guillevin FL, et al. Antineutrophil cytoplasmic antibodies, abnormal angiograms and pathological findings in polyarteritis nodosa and Churg-Strauss syndrome: indications for the classification of vasculitides of the polyarteritis Nodosa Group. Rheumatology. 1996;35(10):958-64.
24. Kaufman AH, Niles JL, Foster CS. (1994). ANCA Test in Ophthalmic Inflammatory Disease. International Ophthalmology Clinics. 1994;34(3):215-27.
25. Mitsuyama K, et al. Antibody markers in the diagnosis of inflammatory bowel disease. World J Gastroenterol. 2016;22(3): 1304-10.
INFORMAÇÃO DOS AUTORES





Financiamento: Declaram não haver
Conflitos de Interesse: Declaram não haver.
Recebido em:
9 de Janeiro de 2020.
Aceito em:
26 de Março de 2020.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados
 Ler em inglês
Ler em inglês
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket