

Caroline Oliveira Brêtas; Thiago George Cabral Silva; Ledilma Inês Colodetti Zanandrea; Patrícia Grativol Costa Saraiva; Fábio Petersen Saraiva
DOI: 10.17545/eOftalmo/2020.0002
ABSTRACT
Retinal vasculitis represents a real challenge in ophthalmic practice. Eartly diagnosis and treatment are important for good visual prognosis and to avoid complications. The aim of this article is to develop a protocol for investigation of retinal vasculitis to facilitate diagnosis and management.
Keywords: Retinal vasculitis; Uveitis; Clinical protocols.
RESUMO
Vasculites retinianas representam um verdadeiro desafio na prática oftalmológica. O diagnóstico precoce e tratamento adequados são de extrema importância para obter bom prognóstico visual e evitar complicações. O objetivo deste artigo é elaborar um protocolo para investigação de vasculites retinianas para facilitar o diagnóstico e conduta.
Palavras-chave: Vasculite retiniana; Uveíte; Protocolos clínicos.
INTRODUCTION
Retinal vasculitis are inflammatory changes in the retinal vessels that may be associated with a primary eye condition or a systemic condition, whether idiopathic, inflammatory, immunomediated, infectious, or malignant. The pathogenesis of retinal vasculitis is presumably an autoimmune phenomenon, with evidence of the presence of CD4+ T lymphocytes inside or around the blood vessels(1). Its estimated incidence is 1-2 new cases per 100,000 people in the United States of America(2). Bilateral is its most common form(3).
Vasculitis may present as intraretinal hemorrhage, cotton wool spots, vascular sheathing, occlusion, or leakage(4). Furthermore, there may or may not be a predominance of arteriolar or venular involvement, and this is an important fact during etiological investigation(5). Possible complications include macular edema, neovascularization, vitreous hemorrhage, retinal detachment, epiretinal membrane, and neovascular glaucoma(5) (Figure 12).
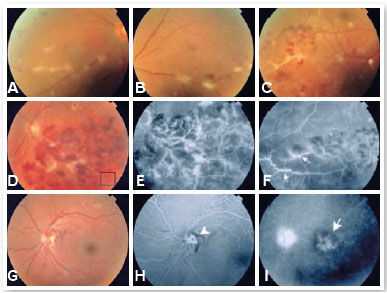
Approximately 14.9% of patients with uveitis present evidence of retinal vasculitis of different etiologies according to Rosenbaum et al.(6). The association with Behçet’s disease is common, whereas ankylosing spondylitis and juvenile idiopathic arthritis are rare(6). In the case of rheumatic diseases, ophthalmologic assessment is important, considering that retinal vasculitis can be asymptomatic, particularly if early-onset or peripheral(7). In addition, they can be the first manifestation of a systemic condition. Early diagnosis is key to a successful treatment and prognosis(7).
METHODS
This study presents a non-systematic literature review. For the bibliographic survey, articles were searched in the PubMed and SciELO databases. The descriptors used were: Retinal Vasculitis; Uveitis; Clinical Protocols.
LITERATURE REVIEW
Epidemiology and classification
Regarding etiology, retinal vasculitis can be: idiopathic, a primary eye condition, or related to a systemic condition, infection, occlusive disease or tumor (masquerade syndrome)(1).
Idiopathic retinal vasculitis, aneurysms, and neuroretinitis (IRVAN)
Described in 1995 by Chang et al., retinal vasculitis is rare and of unknown etiology. It most commonly affects young, healthy, and female patients. There is no racial predilection. Authors have described an association with systemic vasculitis presenting perinuclear anti-neutrophil cytoplasmic antibodies (p-ANCA)(8,9).
The diagnosis includes three major criteria (retinal vasculitis, aneurysmal dilations at arterial bifurcations, and neuroretinitis) and three minor criteria (peripheral capillary non-perfusion, neovascularization, and macular exudation)(8). Patients present low visual acuity in one or both eyes and vitreitis of varying intensity. Arterioles have aneurysms that extend from the first bifurcations to mid-periphery and are commonly found in the optical disc. Lipid exudation is pronounced and typically found in the peripapillary region and can extend to the macula, causing a macular star(10).
Primary eye conditions
Primary eye conditions include Eales disease, pars planitis or intermediate uveitis, and birdshot retinochoroidopathy. Less common conditions worth mentioning are Vogt-Koyanagi-Harada disease (VKH) and sympathetic ophthalmia(1).
Eales disease is an idiopathic retinal occlusive vasculopathy that bilaterally affects healthy young individuals of 20-45 years of age. Periphlebitis and retinal peripheral capillary non-perfusion are observed mainly in the superior temporal quadrant. Its etiology is unknown and association with tuberculin hypersensitivity has been reported. Patients with peripheral neovascularization without any other specific cause are diagnosed with Eales disease(11). Its complications comprise vitreous hemorrhage, tractional or rhegmatogenous retinal detachment, rubeosis iridis, secondary glaucoma, and cataracts(11). Treatment includes ischemic retinal photocoagulation and use of systemic or ophthalmic corticosteroids. Prognosis is generally good(11).
Birdshot retinochoroidopathy is a bilateral chronic posterior uveitis characterized by diffuse inflammation of the choroid associated with vitreitis and retinal vasculitis. It is rare, of unknown cause, and associated with human leukocyte antigen (HLA)-A29. It is most common in Caucasian women who are 30-60 years of age. It affects the choroidal stroma, with poorly defined, oval-shaped, cream-colored depigmented lesions at different stages of evolution located in the posterior pole and mid-periphery. It presents minimal anterior chamber reaction, vitreitis, vascular leakage with macular edema, and sometimes optic disc edema(1). When active, lesions are consistent and, after healing, either leave hypopigmented spots or disappear. Treatment includes corticosteroids and immunosuppressants(1).
Systemic conditions
Among the systemic diseases commonly associated with retinal vasculitis, Behçet’s disease, sarcoidosis, systemic lupus erythematosus, and Wegener’s granulomatosis (WG) are noteworthy. Diseases such as multiple sclerosis, polyarteritis nodosa, inflammatory bowel diseases and other rheumatic diseases may also be associated with it(1).
Behçet’s disease is the rheumatic condition most associated with retinal vasculitis(5). It is a multisystem inflammatory disease related to HLA-B51. It presents with recurrent episodes of occlusive vasculitis affecting mainly small veins and manifests itself with oral and genital ulcers, skin lesions, and eye inflammation. This disease is more serious in men. Ocular manifestations are present in 50%-85% of cases and bilateral in 75%-94%, with eyes being the main organs that are involved. Retinal vasculitis may present vascular sheathing and occlusions, edema, exudates, hemorrhages and infiltrates commonly associated with vitreitis. There may be iridocyclitis and hypopyon(12).
Sarcoidosis is a chronic multisystem inflammatory disease of unknown etiology characterized histologically by the formation of non-necrotizing granulomas. Eye inflammation is usually bilateral and often granulomatous. It affects young adults who are 20-40 years of age. This systemic condition predominates in women; however, eye involvement does not show predilection for sex. It is mainly a periphlebitis with discontinuity. Changes in the posterior segment include: intermediate uveitis with vitreitis, snowballs and/or snowbanks, peripheral vasculitis (perivenular sheathing, perivenous exudates as candle wax drippings), choroidal or optic disc granulomas, preretinal nodules (Landers’ sign), and small nummular atrophic scars on the peripheral retina(13).
Polyarteritis nodosa is an atypical disease characterized by necrotizing vasculitis of small- and medium-sized arteries, with a particular predilection for renal and visceral arteries. It is of great importance because it can involve the posterior ciliary arteries and choroidal vessels, and vasculitis leads to choroidal infarction and secondary retinal detachment(1).
Microscopic polyangiitis (MPA) is a necrotizing vasculitis that affects the microvasculature (arterioles, capillaries, and venules) and rarely medium-sized arteries. It is a rare autoimmune disorder, associated with the presence of anti-neutrophil cytoplasmic antibodies (ANCA), with a predominance of p-ANCA over c-ANCA. Association with central retinal artery occlusion has been reported(14).
Eye involvement in systemic lupus erythematosus (SLE) may reflect the severity and systemic nature of the condition and may be the first symptom reported. The prevalence of lupus retinopathy varies from 3% in outpatients with mild to absent disease to 29% in patients with active disease. In addition, the presence of antiphospholipid antibodies is associated with an increased prevalence of vaso-occlusive retinopathy and vascular occlusions, mainly in the central retinal artery. Although it has a predilection for arteries, associated central retinal vein occlusion has been reported(7,15).
Retinal involvement in WG is unusual (5%-12%), presenting more commonly as hemorrhages of the peripheral retina. It is associated with arterial and venous abnormality, hemorrhages, edema, and cotton wool spots, and may also affect the optic nerve and feature disc edema(1,7).
In patients with rheumatoid arthritis, a 16% prevalence of ophthalmological manifestations has been reported. The most common are scleritis, necrotizing scleritis and PUK. The last two indicate the presence of active systemic vasculitis and increased mortality. Retinal vasculitis and choroiditis are rare events(7).
In the case of spondyloarthropathies, involvement of the anterior segment of the eye is more common, whereas posterior segment involvement can occur in up to 20% of the patients. The presence of HLA-B27 in a patient with uveitis poses a higher risk of complications compared with those with idiopathic anterior uveitis(7,16).
Crohn’s disease is a granulomatous transmural inflammatory bowel disease characterized by a predilection for the distal ileum. It is the most common inflammatory bowel disease associated with extraintestinal manifestations. Approximately 5%-10% of patients have eye complications such as keratitis, episcleritis, iridocyclitis and macular edema. There are reports of association with central retinal artery occlusion and optic nerve vasculitis(17).
Infectious diseases
Infectious causes can be divided into: bacterial, such as syphilis and tuberculosis; viral, such as herpes, human immunodeficiency virus (HIV), and cytomegalovirus (CMV); and parasitic, such as toxocariasis and toxoplasmosis. In cases of immunosuppressed patients with systemic vasculitis who present retinal vasculitis, the most common cause is an infection rather than the underlying disease(5).
Ocular syphilis can manifest in different ways and has thus been called “the great imitator”. Its main form is panuveitis, but it can also occur in other forms such as keratitis, iritis, optic neuritis, macular edema, vitreitis, and retinitis. A rare and severe presentation is central artery occlusion(18).
In the case of vasculitis related to a masquerade syndrome, it is important to consider non-Hodgkin lymphoma of the central nervous system, leukemia, melanoma, intraocular metastasis of carcinomas, retinoblastoma and systemic embolization of cholesterol plaques(19,20).
Below are the main diagnoses according to etiological classification(1):
Etiological classification:
• Idiopathic
– IRVAN syndrome
• Primary eye diseases
– Eales disease
– Birdshot retinochoroidopathy
– Intermediate uveitis
– Vogt-Koyanagi-Harada syndrome
– Sympathetic ophthalmia
– Acute multifocal retinal vasculitis
• Systemic conditions
– Behçet’s disease
– Sarcoidosis
– Multiple sclerosis
– WG
– SLE
– Polyarteritis nodosa
– Microscopic polyangiitis
– Crohn’s disease
– Ankylosing spondylitis
– Buerger’s disease
– Relapsing polychondritis
– Antiphospholipid syndrome
– Churg-Strauss Syndrome
– Polymyositis
– Rheumatoid arthritis
– Dermatomyositis
– Takayasu arteritis
• Infectious causes
– Bacterial: syphilis, tuberculosis, Lyme disease, Whipple’s disease, brucellosis, cat scratch disease, rickettsiosis
– Viral: herpes simplex, varicella zoster, CMV, HIV, human T-lymphotropic virus (HTLV), hepatitis, Epstein-Barr virus, dengue virus
– Parasitic: toxocariasis and toxoplasmosis
– Fungal endophthalmitis
• Occlusive diseases
– Thrombophilia
– Hemoglobinopathies
• Masquerade syndrome
– Tumors: primary central nervous system lymphoma, vitreoretinal lymphoma, acute leukemia, cancer-associated retinopathy
– Metastases
CLINICAL MANIFESTATIONS
Vasculitis may be asymptomatic at first, particularly when the vascular changes are developed in the peripheral retina(5).
Clinical symptoms include: painless vision blurring, changes in color perception, metamorphopsia, floaters, and scotomas. Less commonly, there may be pain and decline in the ability to distinguish colors. Signs and symptoms of systemic involvement may be present, notable of which are: oral, genital, and skin ulcers, arthritis, rash, neurological disease and evidence of embolic disease(1,5,7).
Vascular sheathing is the most common feature present in fundus biomicroscopy, in addition to cotton wool spots, vitreitis of varying intensity, snowballs, vitreous detachment, hemorrhages, macular edema, neovascularization and anterior uveitis. It should be noted that not all cases of vascular sheathing are caused by vasculitis, congenital vascular sheathing being an example. Its occurrence is common and normally present within the two diameters of the optic nerve head, associated with the vein, and may be related to persistent hyaloid vasculature. Vitreous haze translates the breakdown of the blood-retinal barrier, but it is not specific to an inflammatory process; in contrast, vitreous cellularity is a sign of an inflammatory condition(1).
Choroidal inflammation suggests sarcoidosis, birdshot retinochoroidopathy or ocular histoplasmosis syndrome(1).
Late alterations secondary to occlusion and remodeling include telangiectasia, microaneurysms, central vein or branch retinal vein occlusion and ischemia-induced neovascularization; sequelae may follow, such as vitreous hemorrhage, tractional retinal detachment, rubeosis iridis and neovascular glaucoma(1).
According to clinical manifestations, retinal vasculitis can also be classified into(1):
• Infectious or non-infectious
• Arterial, venous, or mixed
• Unilateral or bilateral
• Posterior pole or peripheral retina
• Focal, segmental or diffuse
• Occlusive or non-occlusive
Medical history
Medical history should be considered carefully in order to identify risk factors and personal or family health history that may be useful to understanding the etiology.
Next, we discuss about the important data to be collected for medical history, correlating them with the possible etiology(1,21).
• Jaw claudication, scalp hypersensitivity, polymyalgia rheumatica, palpable temporal artery
– Giant cell arteritis (Takayasu arteritis)
• Repeated gestational losses, recurrent thrombosis
– Thrombophilia
– Antiphospholipid syndrome
– Hemoglobinopathies
• Joint symptoms
– Ankylosing spondylitis
– SLE
– Polyarteritis nodosa
– Relapsing polychondritis
– Buerger’s disease
– Rheumatoid arthritis
• Neurological symptoms
– Multiple sclerosis
• Intestinal symptoms
– Crohn’s disease or ulcerative colitis
• Oral and genital ulcers, pathergy test
– Behçet’s disease
• Previous or suspected malignant neoplasm
– Masquerade syndrome
• Previous blood transfusions
– Hepatitis B and C
– HIV
– HTLV
In addition to these data, it is also important to investigate the history of smoking and oral contraceptive use, which may be associated with occlusive syndromes.
Ophthalmological examination
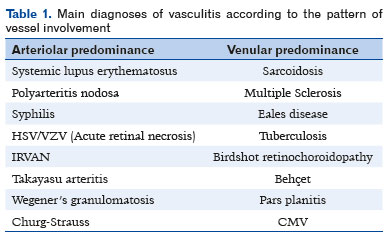
The pattern of vascular involvement and location can help in the diagnosis as different etiologies usually manifest tropism for different types of blood vessels, such as SLE and WG, which affect arteries, and Behçet’s disease, sarcoidosis and multiple sclerosis, which have a preference for veins(5). The main diagnoses according to the affected vessel pattern can be found in table 1(1).
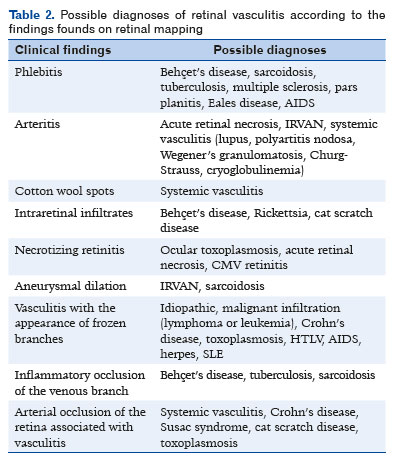
Other ophthalmological alterations observed upon retinal mapping may also assist in diagnosing the cause of retinal vasculitis (Table 2)(1).
Additional tests and examinations
Vasculitis can be detected clinically or by additional examinations, such as fluorescein angiography (FA), in cases with minimal or subclinical signs(7). FA shows the presence of stagnant dye inside the vessel or extravasation through the vessel wall(1,5). Dye leakage leads to progressive hyperfluorescence of the vessel wall in the active disease and evidences increased vascular permeability. In the case of optic nerve vasculitis, nerve head edema and ischemic optic neuropathy are observed(7). Cotton wool spots are seen in AF as focal non-perfusion areas. Optical coherence tomography (OCT) is useful to detect and confirm the presence of subretinal fluid in cases featuring macular edema(1). Biopsy is rarely performed, in view of the potential damage to the retina(1,5). Laboratory tests are fundamental for etiological diagnosis. Therefore, when faced with a new case, considering the most common causes of retinal vasculitis and in the absence of an already defined underlying disease, the following tests and exams are recommended:
• Retinography
• OCT
• FA
• General tests and exams
– Blood pressure measurement
– Complete blood count
– Platelet count
– Fasting blood glucose
– Urea and creatinine
– Urine test
• Infectious diseases serologies
– HIV 1 and 2
– HTLV
– Venereal disease research laboratory test
– Fluorescent treponemal antibody absorption test
– Hepatitis A, B, and C
– Toxoplasmosis IgM and IgG antibodies
– CMV IgM and IgG antibodies
– Herpes IgM and IgG antibodies
– Toxocariasis IgM and IgG antibodies
– Tuberculin (purified protein derivative)
– Bartonella henselae
• Serologies for autoimmune diseases
– Erythrocyte sedimentation rate, C-reactive protein
– ANA, c-ANCA, p-ANCA, anti-Sm, anti-dsDNA
– Anti-Saccharomyces cerevisiae antibodies (ASCA)
– Rheumatoid factor (RF), anti-cyclic citrullinated peptide (CCP)
– HLA (HLA-B27/HLA-A29/HLA-B51)
– Lupus anticoagulants, IgM/IgG anticardiolipin antibodies, antiphospholipid antibodies C3, C4, CH50
– Angiotensin-converting enzyme (ACE), lysozyme
In case no diagnostic confirmation is achieved by these examinations or depending on data from the medical history, the investigation may be furthered with:
• Colonoscopy and endoscopy
– History of bowel symptoms (Crohn’s disease and ulcerative colitis)(17)
• Nuclear magnetic resonance imaging of the brain (and, if necessary, of the cervical, thoracic, and lumbar spine) and cerebrospinal fluid analysis(1)
– History of neurologic deficit, increased IgG in the cerebrospinal fluid (multiple sclerosis)(1)
• Temporal artery biopsy(20)
– Jaw claudication, polymyalgia rheumatica, palpable temporal artery, scalp hypersensitivity (temporal arteritis)
• Chest X-ray and chest computed tomography
– Bilateral hilar lymphadenopathy, paratracheal or parenchymal pulmonary infiltrates, fibrosis (sarcoidosis)(1,13)
• Mediastinal lymphadenopathy, primary cavitary lesion, calcified tuberculomas and pulmonary infiltrates, diffuse micronodules (tuberculosis)(1,22)
– Systemic and local tumors and secondary implantation (masquerade syndrome)(19)
• Angiography, computed tomography angiography, or magnetic resonance angiography
– Aneurysms and microaneurysms in medium-sized arteries, predilection for renal and visceral arteries (polyarteritis nodosa)(23)
• Ocular ultrasound
– Intraocular masses (tumors)(1,19)
• Vessel biopsies(1,21)
• Hb electrophoresis(21)
– Hemoglobinopathies
• Bilirubin level(1)
– Hepatic changes, hemolysis (anemia, hepatitis)
• Coagulation test (factors VII, IX, and XI), proteins C and S, factor V Leiden, prothrombin gene mutation, homocysteine(1)
• Investigation of thrombophilia(1)
• Serologies for dengue, rickettsiosis, Lyme disease, cat scratch disease(1)
• Aqueous humor polymerase chain reaction(1)
– Vitreous biopsy (cases with suspected malignancy)(1,19)
When a specific infectious serology is positive, a presumed diagnosis can be made. Specific immunological tests are usually positive in only certain conditions. The following conditions are associated with these respective positive tests:
• SLE: decreased ANA, anti-Sm, anti-dsDNA, CH50, HLA-DR3, C3, and C4(13,15,21)
• Antiphospholipid syndrome: lupus anticoagulants, anticardiolipin antibodies, antiphospholipid antibodies(15)
• Behçet’s: pathergy test, HLA-B51(12)
• Polyarteritis nodosa: hepatitis serology (15%-30% of patients have a hepatitis B virus infection, 5% have hepatitis C), c-ANCA, p-ANCA(23,24)
• Sarcoidosis: ACE, lysozyme, increased serum calcium(13)
• Rheumatoid arthritis: RF, anti-CCP(21)
• Ankylosing spondylitis: HLA-B27(7)
• Birdshot: HLA-A29(21)
• WG: c-ANCA, p-ANCA(24)
• Churg-Strauss syndrome: c-ANCA, p-ANCA(23,24)
• Crohn’s disease: ASCA(25)
• Ulcerative colitis: p-ANCA(25)
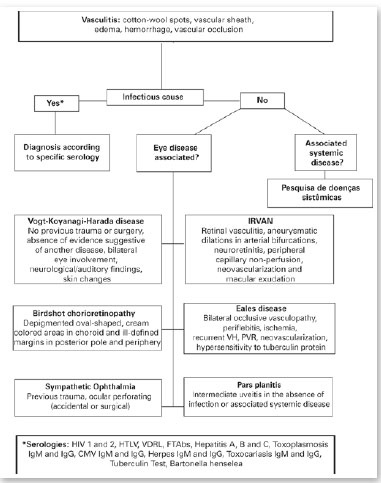
A flowchart (A) in the annex presents a proposed conduct for new cases of retinal vasculitis.
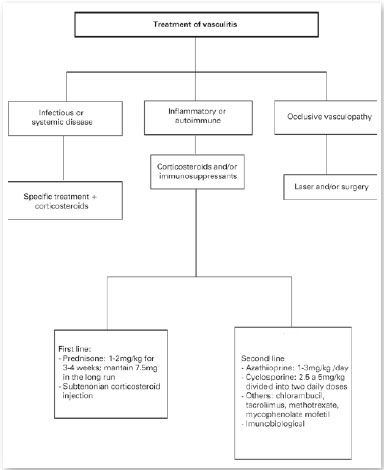
TREATMENT
Treatment varies according to causes, signs, and symptoms. The objective is to suppress intraocular inflammation in order to prevent vision loss and long-term complications, as well as to maintain remission and prevent relapses. Therapy may not be required in cases with minimal disease activity and a 20/40 visual acuity or better. In cases of mild inflammation, good visual acuity and absence of extensive retinal involvement, the choice may be monitoring or using topical steroids or sub-Tenon’s steroid injections.
In the most severe cases, with significant loss of visual acuity, macular edema, and extensive retinal involvement, the use of systemic corticosteroid therapy (oral or intravenous) is indicated. Once the disease is controlled, medication should be gradually reduced. Immunosuppressant drugs such as azathioprine, methotrexate and cyclosporine can be tried in refractory cases. Vitrectomy and laser photocoagulation are indicated in cases of persistent neovascularization, vitreous hemorrhage or glaucoma(1).
Infectious causes should be treated considering the etiological agent. In cases secondary to Behçet’s disease, anti-TNF monoclonal antibodies or interferon-alpha are chosen. Occlusive vasculitis, such as antiphospholipid syndrome, is treated with antiplatelet and/or anticoagulant therapy.
In most cases, treatment is performed with local or systemic corticosteroids in combination or without immunosuppressants and subsequently immunobiological therapy. Corticosteroids are more rapidly effective, whereas immunosuppressants may take up to 3 months to take effect(5).
Sub-Tenon’s steroid injections are useful in unilateral or severe cases. The periocular route prevents the systemic side effects of the oral route, despite the risk of increased intraocular pressure, cataracts or inadvertent globe perforation. Clinical improvement is observed within 2-3 weeks after the procedure and effects can last up to 3 months.
Oral corticosteroid therapy is typically used in moderate to severe bilateral cases with significant loss of visual acuity. The recommended dose is 1-2mg/kg. Some authors recommend the high-dose regimen for 3-4 weeks, whereas others advocate a 50% dose reduction every 8 days followed by 1 month of a reduced dose. If there is no recurrence, the corticosteroids may be maintained for long term at a dose of 7.5mg/day.
Immunosuppressants should be considered in insufficiently or non-responsive cases and when there are side effects to corticosteroids. The agents that can be used are: azathioprine, cyclophosphamide, cyclosporine, methotrexate, chlorambucil, mycophenolate mofetil, and tacrolimus. Of these, azathioprine and cyclosporine are the most used.
➞ Cyclosporine: 2.5-5mg/kg/day divided into two daily doses
➞ Azathioprine : 1-3mg/kg/day
The use of anti-vascular endothelial growth factor antibodies as well as corticosteroids in the form of intravitreal injections can be attempted to manage complications such as macular edema.
The flowchart for vasculitis treatment is in the annex (Flowchart B).
PROGNOSIS
The prognosis for vision is variable. Some present a benign progression with good response to therapy, preserving visual acuity. However, more resistant conditions, even with immunosuppressants, may progress to functional visual loss, resulting in a guarded prognosis for vision.
REFERENCES
1. Siqueira RC, Oréfice F. Vasculites da Retina. In: Oréfice, F. et al. Uveítes. Série Oftalmologia Brasileira. 4ª edição, Rio de Janeiro, Cultura Médica, 2016. Cap.11, p. 105-123.
2. Hughes EH, Dick AD. The pathology and pathogenesis of retinal vasculitis. Neuropathol Appl Neurobiol. 2003;29(4):325-40.
3. Ali A, Ku JH, Suler EB, el al. The course of retinal vasculitis. Br J Ophthalmol. 2014;98(6):785-9.
4. Tahreem AM, et al. Clinical Features and incidence rates of ocular complications in patients with retinal vasculitis. Am J Ophthalmol. 2017;179:171-8.
5. Rosenbauma JT, Sibley CH, Lin P. Retinal Vasculitis. Curr Opin Rheumatol. 2016;28(3):228-35.
6. Rosenbauma JT, Ku J. Ali, A., et al. Patients with Retinal Vasculitis rarely suffer from systemic vasculitis. Semin Arthritis Rheum. 2012;41(6):859-65.
7. Androudi S, Dastiridou A, Symeonidis C, et al. Retinal vasculitis in rheumatic diseases: na unseen burden. Clin Rheumatol. 2013; 32(1):7-13.
8. Chang TS, Aylward GW, Davis JL, et al. Idiopathic retinal vasculitis, aneurysms, and neuro-retinitis: Retinal Vasculitis Study. Ophthalmology. 1995;102(7):1089-97.
9. Soheilian M, Nourinia R, Tavallali A, et al. Idiopathic retinal vasculitis, aneurysms, and neuroretinitis syndrome associated with positive perinuclear antineutrophil cytoplasmic antibody (P-ANCA). Retin Cases Brief Rep. 2010;4:198-201.
10. Agarwal A. Idiopathic retinal vasculitis, aneurysms, and neuroretinopathy (IRVAN). In: Gass’ Atlas of Macular Diseases. 5ª edição. Philadelphia: Saunders: 2012, p.534-538.
11. Jr Mathura JR, Bearelly S, Jampol LM. Retinopatias Proliferativas. In: Yanoff, M.; Ducker J.S.; Retina e Vítreo. 4ª edição, Rio de Janeiro, Elsevier, 2017. Cap. 26, p.152-156.
12. Oréfice F, Diligente FT, Oréfice JL. Doença de Behçet. In: Oréfice, F. et al. Uveítes. Série Oftalmologia Brasileira. 4ª edição, Rio de Janeiro, Cultura Médica, 2016. Cap.33, p. 369-377.
13. Oréfice F, Diligente FT, Cattan JM. Sarcoidose. In: Oréfice, F. et al. Uveítes. Série Oftalmologia Brasileira. 4ª edição, Rio de Janeiro, Cultura Médica, 2016. Cap.32, p. 361-367
14. Domingues CG, Capitanio CF, Abreu LB, et al. Central retinal artery occlusion in patient with microscopic polyangiitis. Rev Bras Oftalmol. 2015;74(6):386-9.
15. Seth G, Chengappa KG, Misra DP, et al. Lupus retinopathy: a marker of active systemic lupus erythematosus. Rheumatol Int. 2018;38(8):1495-501.
16. Power WJ, Rodriguez A, Pedroza-Seres M. Outcomes in anterior uveitis associated with the HLA-B27 haplotype. Ophthalmology. 1998;105(9):1646-51.
17. Coussa RG, Ali-Ridha A, Vila N. Simultaneous central retinal artery occlusion and optic nerve vasculitis in Crohn disease. Am J Ophthalmol Case Rep. 2017;5:11-5.
18. Khan MS, Kuruppu DK, Popli TA, et al. Unilateral optic neuritis and central retinal vasculitis due to ocular syphilis. Retin Cases Brief Rep. 2017
19. Oréfice F, Diligente FT, Cattan J M. Síndromes Mascaradas. In: Oréfice, F. et al. Uveítes. Série Oftalmologia Brasileira. 4ª edição, Rio de Janeiro, Cultura Médica, 2016. Cap.32, p. 425-438.
20. Jacobson DM. Systemic Cholesterol Microembolization Syndrome Masquerading as Giant Cell Arteritis. Survey of Ophthalmology. 1991;36(1):23-7.
21. Agarwal A, et al. Retinal Vasculitis. EyeWiki, São Francisco, CA, 16 de dez. 2019. Disponível em: <https://eyewiki.aao.org/Retinal_Vasculitis> Acesso em: 30 de fev de 2020.
22. Castro MC, Oréfice F. Tuberculose. In: Oréfice, F. et al. Uveítes. Série Oftalmologia Brasileira. 4ª edição, Rio de Janeiro, Cultura Médica, 2016. Cap.23, p. 233-253.
23. Guillevin FL, et al. Antineutrophil cytoplasmic antibodies, abnormal angiograms and pathological findings in polyarteritis nodosa and Churg-Strauss syndrome: indications for the classification of vasculitides of the polyarteritis Nodosa Group. Rheumatology. 1996;35(10):958-64.
24. Kaufman AH, Niles JL, Foster CS. (1994). ANCA Test in Ophthalmic Inflammatory Disease. International Ophthalmology Clinics. 1994;34(3):215-27.
25. Mitsuyama K, et al. Antibody markers in the diagnosis of inflammatory bowel disease. World J Gastroenterol. 2016;22(3): 1304-10.
AUTHOR’S INFORMATION





Funding: No specific financial support was available for this study
Disclosure of potential conflicts of interest: None of the authors have any potential conflict of interest to disclose
Received on:
January 9, 2020.
Accepted on:
March 26, 2020.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados
 Ler em português
Ler em português
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket