

Maria Isabel Lynch Gaete1; Ana Carolina Visco de Almeida2; Talita Raquel Sampaio Patriota3; Diogo Wagner Galindo Vaz Veras de Queiroz4; Jonielly Costa Vasconcelos de Santana5
DOI: 10.17545/eoftalmo/2018.0015
RESUMO
A miopatia mitocondrial ou Síndrome de Kearns-Sayre é uma doença rara que contempla a tríade: indivíduos na segunda década de vida, oftalmoplegia externa progressiva crônica e degeneração pigmentária da retina. Neste relato, descreve-se o caso de um paciente que há 6 anos iniciou um quadro de ptose palpebral bilateral progressiva, hiperemia ocular e lacrimejamento com piora ao redor de 1 ano, com passado de correção de ptose em duas ocasiões em outro serviço, evoluindo com ceratite de exposição. O caso apresentado aponta a necessidade de cautela em relação à indicação cirúrgica da ptose palpebral, pois a ausência do Reflexo de Bell decorrente da Miopatia Mitocondrial e a diminuição da força muscular após correção cirúrgica da ptose propicia uma exposição corneana, com insuficiente lubrificação e complicações possíveis como ceratite de exposição.
Palavras-chave: Miopatias mitocondriais; Síndrome de Kearns-Sayre; Oftalmoplegia externa progressiva crônica.
ABSTRACT
Mitochondrial myopathy or Kearns–Sayre syndrome is a rare disease with a typical triad: (1) onset at <20 years of age, (2) chronic progressive external ophthalmoplegia, and (3) pigmentary degeneration of the retina. Herein, we describe the case of a patient presenting with bilateral progressive ptosis, ocular hyperemia, and lacrimation for 6 years, which had been worsening for approximately 1 year, and who had a history of ptosis correction on two occasions at another institution, progressing toward exposure keratitis. This case highlights the need for caution when indicating ptosis correction to patients because the absence of Bell’s phenomenon due to mitochondrial myopathy and decreased muscle strength following this surgical treatment cause corneal exposure with insufficient lubrication and possible complications, such as exposure keratitis.
Keywords: Mitochondrial Myopathies; Kearns-Sayre Syndrome; Ophthalmoplegia, Chronic Progressive External.
RESUMEN
La miopatía mitocondrial o Síndrome de Kearns-Sayre es una enfermedad rara que contempla la siguiente tríada: individuos en la segunda década de vida, oftalmoplegia externa progresiva crónica y degeneración pigmentaria de la retina. En este relato, se describe el caso de un paciente que hace 6 años inició un cuadro de ptosis palpebral bilateral progresiva, hiperemia ocular y lagrimeo con empeoramiento alrededor de 1 año, con un pasado de corrección de ptosis en dos ocasiones en otro servicio, evolucionando para queratitis de exposición. El caso presentado señala la necesidad de cautela respecto a la indicación quirúrgica de la ptosis palpebral, dado que la ausencia del Reflejo de Bell consecuente de la Miopatía Mitocondrial y la disminución de la fuerza muscular tras corrección quirúrgica de la ptosis propician una exposición corneal, con insuficiente lubricación y posibles complicaciones tales como queratitis de exposición.
Palabras-clave: Miopatías Mitocondriales; Síndrome de Kearns-Sayre; Oftalmoplejía Externa Progresiva Crónica.
INTRODUÇÃO
A primeira descrição da doença ocorreu em 1958 pelos autores Kearns e Sayre sob a forma de um relato de caso de 2 casos de pacientes jovens apresentando alterações oculares associadas a distúrbios de condução cardíaca, sob a forma de bloqueios átrio-ventriculares, as quais podem ser fatais. Em 1965 foi reconhecida como uma síndrome e passou a ser denominada Síndrome de Kearns-Sayre (SKS). Trata-se de uma entidade rara, na qual as manifestações clínicas surgem por volta dos 20 anos de idade, incluindo alterações oftalmológicas que vão desde oftalmoplegia externa crônica progressiva, alterações retinianas até ceratopatia de exposição. Faz-se necessária, portanto, a investigação etiológica para o diagnóstico precoce e manejo adequado das situações de risco, assim como a cautela na indicação da correção de ptose nos pacientes portadores da síndrome.
MÉTODOS
Estudo descritivo tipo relato de caso. Dados obtidos da anamnese e exames complementares do paciente atendido no Serviço Oftalmológico de Pernambuco- SEOPE.
RESULTADOS
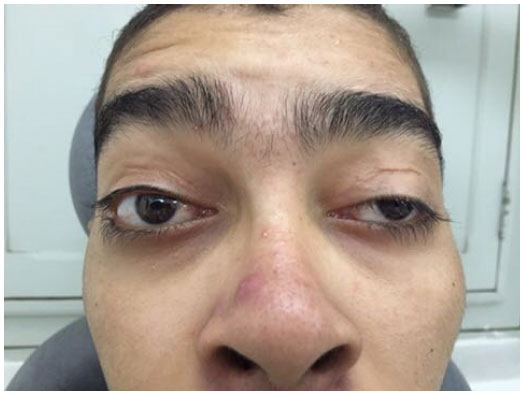
JCS, 20 anos, masculino, compareceu ao Serviço Oftalmológico de Pernambuco (SEOPE) com história de correção cirúrgica de ptose bilateral, em outro serviço, duas vezes há 7 anos. Relatou retorno da ptose palpebral bilateral progressiva há 6 anos, com hiperemia ocular e lacrimejamento, com piora do quadro há 1 ano. À inspeção, observou-se ptose assimétrica com fenda palpebral de 7 mm em olho direito (OD) e do olho esquerdo (OE) de 5 mm (Figura 1). A função do músculo levantador da pálpebra do OD era 2 mm e do OE 5 mm. Distância margem reflexo OD +1 e OE foi zero. O exame de motilidade ocular extrínseca revelou importante limitação muscular no teste de versões e ducções. Ausência do reflexo de Bell. Acuidade visual com correção 0,5 (AO). À biomicroscopia: conjuntiva calma, córnea com opacidade corneana paracentral inferior circular, fluoresceína positiva em OD. Em OE opacidade paracentral inferior linear, fluoresceína negativa. Tonometria: 10 mmHg AO. Fundoscopia: discos ópticos sem alterações, vasos normais, retina com aspecto em “sal e pimenta” em equador e periferia.
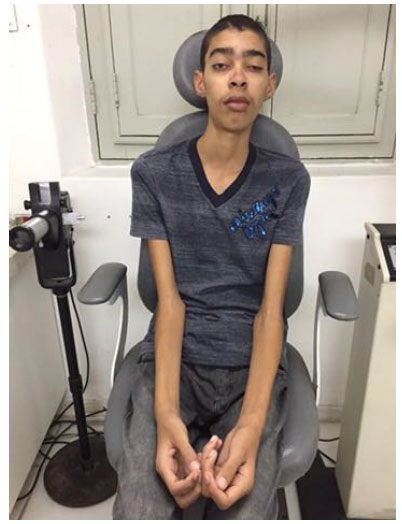
Ao exame clínico sistêmico: pressão arterial 110 X 80 mmHg, hipotrofia muscular generalizada e membros apendiculares alongados (Figura 2). Exames endocrinológico e laboratoriais: T4 livre e TSH normais. CPK elevado (322 U.I./L) LDH elevado (252 mg/dl), aldolase normal. Audiometria: perda auditiva sensório neural bilateral de grau moderado a direita e severa e esquerda. ECG: ritmo sinusal, arritmia supraventricular de baixa incidência, bloqueio de ramo direito fixo. RM de Crânio: restrição à difusão nas áreas afetadas da substância branca subcortical dos lobos occipitais e globos pálidos. Sugestivo de doença metabólica. Eletroforese no liquor: pesquisa de bandas oligoclonais ausentes. Cariótipo: 46 XY (masculino, sem anormalidades). Biópsia Muscular: fibras musculares com aspecto “granulado”. Na reação do Gomori Modificado: presença de fibras rasgada. Presença de fibras COX negativo. Presença de fibras “ragged blue” no SDH. Alterações musculares com padrão de miopatia mitocondrial.
DISCUSSÃO
A Doença de Kearns-Sayre é rara. Herança heterogênea, tendo sido observado vários tipos. Decorre de deleções no DNAmt, das quais a maioria é esporádica e acredita-se que aconteçam como mutações de células germinativas ou muito precoces no desenvolvimento embrionário. É uma doença multissistêmica e apresenta uma tríade de características que inclui: início em indivíduos com idade menor que 20 anos, oftalmoplegia externa crônica progressiva e degeneração pigmentária da retina. Outros sintomas são: fraqueza muscular, diminuição crônica e progressiva dos movimentos dos olhos e ptose, disfagia, disfunção do SNC, ataxia, demência, encefalopatia ou ambos, surdez, cegueira noturna, síncope e defeitos na condução cardíaca 1,2,3. A ptose palpebral geralmente é o primeiro sinal clínico. Usualmente bilateral e simétrica e com a progressão o paciente pode adotar uma posição de elevação da cabeça e queixo 4,5. A diminuição da motilidade pode ser despercebida até se tornar severa. Pode haver queixa de hiperemia ocular devido a ceratopatia de exposição causada pela paralisia de Bell 6. Na Síndrome de Kearns-Sayre não há achados ao nascimento, porém, antes dos vinte anos aparece, geralmente, a oftalmoplegia externa progressiva e a retinopatia pigmentar, com aspecto em sal e pimenta. O paciente pode apresentar defeitos da condução cardíaca com evolução, muitas vezes, para morte súbita 7. O diagnóstico é fornecido por biópsia muscular 8. O tratamento deve ser multidisciplinar com prognóstico reservado e a desordem geralmente é progressiva.
REFERÊNCIAS
1. Nonaka I. Mitochondrial disease. Curr Opin Neurol Neurosurg. 1992;5(5):622-32.
2. Souza CFM. Doenças da cadeia respiratória mitocondrial: um estudo clínico, bioquímico, histológico e molecular [Dissertação de mestrado]. Porto Alegre: Universidade Federal do Rio Grande do Sul (UFRGS); 2001. 100 p.
3. Schmitz K, Lins H, Behrens-Baumann W. Bilateral spontaneous corneal perforation associated with complete external ophthalmoplegia in mitochondrial myopathy (Kearns-Sayre syndrome). Cornea. 2003;22(3):267-70.
4. Cohen JM, Waiss B. Combination ptosis crutch and moisture chamber for management of progressive external ophthalmoplegia. J Am Optom Assoc. 1997;68(10):663-7.
5. Dias-Tosta E. Chronic progressive external ophthalmoplegia: I. Aquantitative histochemical study of skeletal muscles. Arq Neuropsiquiatr. 1988;46(2):133-42.
6. Fraunfelder FT, Roy FH, Randall J. Chronic progressive external ophthalmoplegia. In: Fraunfelder FT, Roy FH. Current Ocular Therapy. 5th ed. Philadelphia: W.B. Saunders; 2000. p. 208-10.
7. Cruz MW, André C, Hahn MD, Mattos JP, Maranhão Filho PA, Silva ES, et al. Síndrome de Kearns-Sayre: relato de caso com instalação rápida e documentação anátomo-patológica. Rev Bras Neurol. 1989;25(2):55-60.
8. Consoni Filho E, Oliveira ASB, Schmidt B. Musculatura extra-ocular: um músculo esquelético diferenciado. Arq Bras Oftalmol. 1994;57(6):394-9.





Fonte de financiamento: declaram não haver.
Parecer CEP: não aplicável.
Conflito de interesses: Declaram não haver.
Recebido em:
10 de Abril de 2018.
Aceito em:
7 de Junho de 2018.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados
 Ler em inglês
Ler em inglês
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket