

Alexis Galeno Matos1; Rigoberto Gadelha Chaves2; Marília de Freitas Chaves3; Renata Girão Cavalcante4
DOI: 10.17545/eoftalmo/2018.0008
ABSTRACT
Gaucher disease(GD) is autosomal recessive due to the deficiency of the acid beta glucosidase enzyme caused by mutations in the 1q21 gene, promoting the accumulation of glucosylceramide in cells of the reticuloendothelial system. The main clinical manifestations of the DG are anemia, thrombocytopenia, hepatosplenomegaly, bone alterations and neurological manifestations and they are related to macrophages loaded with non-metabolized fats. The main clinical presentations are non-neuronopathic (DG1), acute neuronopathic (DG2) and subacute neuronopathic (DG3). Other organs, such as the lungs, kidneys and eyes, may be affected. Significant ocular alterations and oculomotor disorders may occur in DG. The treatment of DG is clinical and must be carried out during the life. Although the success of treatment of ocular manifestations is still questionable, ophthalmologic assessment is of great importance for the followup of patients with DG.
Keywords: Gaucher Disease, Glucosylceramide, Macrophages.
RESUMO
A doença de Gaucher DG é autossômica recessiva decorrente da deficiência da enzima beta-glucosidase ácida causada por mutações no gene 1q21, promovendo o acúmulo de glucosilceramida na células do sistema reticuloendotelial. As principais manifestações clínicas da DG são anemia, trombocitopenia, hepatoesplenomegalia, alterações ósseas e manifestações neurológicas, que são relacionadas aos macrófagos carregados de gorduras não metabolizadas. As principais apresentações clínicas são doença não-neuronopática (DG1), neuronopática aguda (DG2) e neuronopática subaguda (DG3). Outros órgãos, como pulmões, rins e olhos, podem ser acometidos. Importantes alterações oculares e distúrbios oculomotores podem ocorrer na DG. O tratamento da DG é clínico e deve ser realizado por toda vida. Embora o sucesso do tratamento das manifestações oculares ainda seja questionável, a avaliação oftalmológica é de grande importância no seguimento dos pacientes com DG.
Palavras-chave: Doença de Gaucher, Glucosilceramida, Macrófagos.
RESUMEN
La enfermedad de Gaucher (EG) es autosómica recesiva consecuente de la deficiencia de la enzima beta-glucosidase ácida causada por mutaciones en el gene 1q21, promoviendo el acúmulo de glucosilceramida en la células del sistema reticuloendotelial. Las principales manifestaciones clínicas de la EG son anemia, trombocitopenia, hepatoesplenomegalia, alteraciones óseas y manifestaciones neurológicas, las cuales están relacionadas a los macrófagos cargados de grasas no metabolizadas. Las principales presentaciones clínicas son enfermedad no neuronopática (EG1), neuronopática aguda (EG2) y neuronopática subaguda (EG3). Otros órganos, como pulmones, riñones y ojos, pueden ser acometidos. Importantes alteraciones oculares y disturbios oculomotores pueden ocurrir en la EG. El tratamiento de la EG es clínico y debe realizarse de manera vitalicia. Aunque el éxito del tratamiento de las manifestaciones oculares aún sea cuestionable, la evaluación oftalmológica es de grande importancia en el seguimiento de los pacientes con EG.
Palabras-clave: Enfermedad de Gaucher; Glucosilceramida; Macrófagos.
Gaucher disease (GD, OMIM #230800, ORPHA355) is a rare genetic disease caused by mutations in the GBA1 gene (GBA; 606463), which is located on chromosome 1 (1q21)1, that sharply reduce the activity of the enzyme glucocerebrosidase (GCase, also known as glucosylceramidase or acid β-glucosidase, EC 4.2.1.25) in the lysosomes. GCase hydrolyzes glucosylceramide (GlcCer) into ceramide and glucose resulting from the degradation of red and white blood cell membranes. Its deficiency, therefore, leads to glucocerebroside accumulation in tissues, especially in mononuclear phagocyte system cells.2
EPIDEMIOLOGY
The available data on the birth incidence of GD in the general population in several countries ranges from 0.39 to 5.80 per 100,000 births, while the birth prevalence ranges from 1.33 to 1.75 per 100,000 births.3 The autosomal recessive inheritance of GD allows the inference that consanguineous marriages and the founder effect of certain mutations in specific populations can lead to the phenotypic manifestations of recessive genes over several generations as verified in the town of Tabuleiro do Norte, State of Ceará, Brazil where the prevalence of the disease is approximately 1:4,000 people.4
CLINICAL MANIFESTATIONS
The clinical manifestations of GD are associated with different actions of the following two types of macrophages: M1 (classically activated macrophages) and M2 (alternatively activated macrophages). M2 macrophages are believed to be involved in the production of cytokines, chemokines, and other molecules including IL-1; IL-6; IL-8; TNF-α (tumor necrosis factor alpha); M-CSF (macrophage colony-stimulating factor); MIP-1; IL-18; IL-10; TGF; CCL- 18; chitotriosidase; and CD14s, being responsible for the disease’s “pseudo- inflammatory” reactions5-7. Macrophages engorged with fat, characterized by fibrillar cytoplasm with a “crumpled paper” appearance and eccentric nuclei, reacting positively to PAS (periodic acid-Schiff) staining, are called “Gaucher cells” (GC).8
More than 300 mutations cause GD and, alone as well as in combination in both alleles, can determine the phenotypic manifestations: type 1 or non- neuronopathic (94%; GD1, OMIM #230800); type 2 or acute neuronopathic (GD2, OMIM #230900), or type 3, also called subacute neuronopathic (GD3, OMIM #231000).9,10
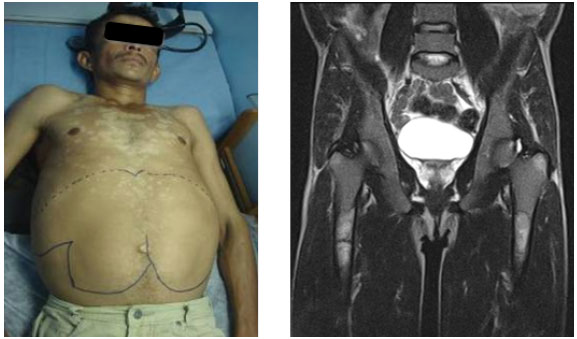
The rarity of the disease and the heterogeneous clinical manifestations hinder the early diagnosis of GD.11 GD1 is characterized by hepatosplenomegaly, anemia with thrombocytopenia, lung diseases, presence of clinical or radiographic evidence of bone diseases, and absence of primary central nervous system diseases (Figure 1).12
GD2 (<5% of cases) has its onset before age 2, with impaired psychomotor development and a fast, progressive course that leads to death between 2 and 4 years12. Patients experience severe neurological impairment, consisting of neck and trunk spasticity (opisthotonus), bulbar signs (especially severe swallowing disorders), and oculomotor nerve palsy (or bilateral strabismus fixus). In addition to these signs, there may also be trismus and hypertonia with pyramidal and possibly extrapyramidal rigidity7.
The GD3 form, also called juvenile or subacute neurological GD, represents only 5% of GD cases, although it can reach up to 33% in some groups of patients13. These patients usually present with the visceral manifestations described in GD1 associated with oculomotor neurological involvement that appear in most cases before the age of 20 years. Some patients have moderate systemic involvement, with horizontal ophthalmoplegia as the only neurological symptom, while others feature more serious forms, with several neurological signs including progressive myoclonus epilepsy (16% of patients), cerebellar ataxia or spasticity (20%–50% of patients), and, in some cases, dementia14,15.
OPHTHALMOLOGICAL MANIFESTATIONS
Ophthalmological changes in GD can manifest in the anterior and posterior segments in addition to affecting eye movements in some instances because of the neurological changes.16-18
CHANGES IN THE ANTERIOR SEGMENT OF THE EYE
Gaucher first described in 1882 a disease that caused enlarged spleen with jaundice and yellowish conjunctiva. According to McNee (1937), in this disease “the eyes show a phenomenon which is so constant as to be almost pathognomonic: a brownish-yellow wedge-shaped thickening or pinguecula appears first on the nasal and then on the temporal side of each eye”.19 The biopsy of the pinguecula showed a degenerated, foamy cytoplasm with no fat particles, similar to that observed in spleen cells in GD cases.19 Recent histopathological studies found pinguecula elastosis.20
The ocular manifestations of GD1 include white deposits scattered throughout the corneal endothelium, anterior chamber angle, ciliary body, and pupillary margin. Corneal involvement is variable and correlates with the type of mutation.21 Bilateral corneal opacity has been observed on three levels using biomicroscopy. They featured evident scattered areas of subepithelial opacity and focal areas of thickening and opacities in the stroma which were greater in the posterior third. The architecture of the corneal stroma is distorted with keratocytes interspersed with multiple small white dots caused by extensive cytoplasmic vacuolation. The deep stroma has a spiculated appearance. Corneal opacities are presumed to result from the accumulation of GlcCer in the keratocytes. Endothelial cell count may be lower than that expected for the patient’s age.22
CHANGES IN THE POSTERIOR SEGMENT OF THE EYE
Fundus abnormalities include perimacular ring-shaped pigmented lesions, typical white retinal lesions that occur because of the preretinal accumulation of glycolipids, macular atrophy, retinal hemorrhages, and edema caused by increased retinal vascular permeability.21,23
Vitreous opacities are the most frequently occurring manifestations in patients with GD and they are present in 3% of cases24. This finding has been noted to be more frequent after splenectomy due to increased glucosylceramide circulation25. Takehisa et al. assessed sphingolipids in a patient with GD and detected ceramide monohexoside accumulation in the vitreous humor using chromatography26.
Shrier et al. described a vascular tortuosity with a spiral pattern similar to that in Fabry disease as well as the presence of an epiretinal membrane. They also reported the presence of GC in the vitreous cavity, along with large quantities of GlcCer25.
The pathophysiology of the GC in the retina remains unclear, but higher levels of circulating GlcCer in unusual systemic sites have been suggested as the cause for these changes. Retinal GlcCer deposition is an end product of the breakdown of myelin, white blood cells, red blood cells, and endothelial cells. The cells in the retina that absorb this substrate are glial cells: Müller cells, astroglia, and especially microglia.27,28 However, it is not clearly understood whether GlcCer accumulation produces functional changes in the retina of patients who are still visually asymptomatic. Visual function impairment and progressive retinal involvement despite enzyme replacement therapy have been described27.
Seidova et al. described preretinal lesions and subclinical abnormalities on the electroretinogram (ERG) which were attributed to the accumulation of storage lipids in glial cells.27 An association between GD and retinitis pigmentosa has also been proposed since the liver disease leads to vitamin A deficiency.29
There are case reports of patients with GD3 presenting white peripapillary and perivascular deposits on the posterior pole of the eye, which are described in the optical coherence tomography (OCT) as hyporeflective preretinal lesions in the vitreoretinal interface associated with localized posterior vitreous detachment (Figure 2)30,31.
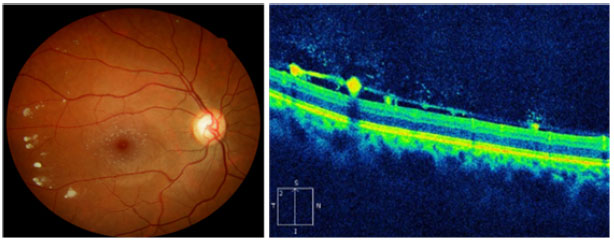
Wang et al. also described yellow-white macular lesions in both eyes. Fluorescein angiography evidenced changes in the retinal pigment epithelium of the right eye macula and irregular areas of hyperfluorescence in the left one.
The retinal vessels showed contrast extravasation, especially in the peripapillary region.21
Central retinal artery branch occlusion has been described in a patient with complicated GD, raising the hypothesis that microthrombi are a part of the etiology of the obstruction of this vessel32. Tractional retinal detachment has also been reported in patients with GD33.
UVEITIS
Uveitis is another change described in GD. GlcCer accumulation is suggested to be involved in some forms of uveitis. An assessment of the sphingolipids found in the vitreous of a patient with GD showed GlcCer accumulation followed by monocyte and macrophage infiltration34. GC have also been found in the choroid of patients with the disease23.
In one study, 2 (0.4%) of 527 patients with GD1 presented with uveitis. One of them presented with bilateral asymmetric panuveitis with keratic precipitates and vitreitis35. Chronic intermediate uveitis with residual snowballs has been reported in patients with GD, with most cases improving with enzyme replacement therapy36. Altoon et al. reported two cases of patients with uveitis presenting different clinical courses and outcomes. Although one patient showed improvement, chronic uveitis persisted in the older patient, despite undergoing enzyme replacement therapy35.
It is important to highlight the association between chronic uveitis resistance to clinical treatment and lymphoma (masquerade syndromes). Increased incidence of B-cell and T-cell malignancies, such as multiple myeloma and chronic lymphocytic leukemia, have been reported in patients with GD. Thus, malignancy should be investigated in cases of treatment-resistant chronic uveitis35.
DECREASED RETINAL NERVE FIBER LAYER (RNFL) THICKNESS
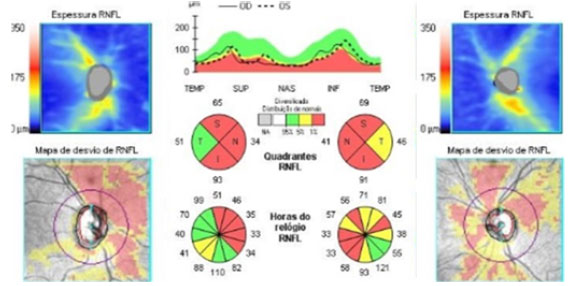
Increased excavation and decreased RNFL, which are suggestive of glaucomatous optic neuropathy, are a rare finding in patients with GD. Some authors attribute the reduction in the nerve fiber layer to the loss of acid β-glucosidase activity, which is associated with oxidative stress and mitochondrial dysfunction37. Matos et al. described a case of GD3 with suspected glaucoma and markedly decreased RNFL (Figure 3)31.
EYE MOVEMENTS
The detailed examination of eye movements is indispensable in clinical investigation, in addition to being useful in the diagnostic elucidation and classification of GD clinical subtypes in relation to neurological involvement38.
In the GD2 and GD3 (neuronopathic) clinical forms, the following oculomotor changes have been described: failure or slowing down of horizontal and downward saccadic eye movements, abnormal vestibuloocular reflex, and altered optokinetic nystagmus39,40.
The most frequently occurring oculomotor change in GD3 is early supranuclear motor control dysfunction, which leads to deficient saccadic eye movements, also called “ocular motor apraxia,” “congenital ocular motor apraxia,” “supranuclear gaze palsy,” “gaze palsy,” and “looping”41. Children can attempt to compensate deficient saccadic eye movements with head thrusting to move their gaze. In older children, head positioning is often replaced with a kinetic blinking strategy.
The ocular motility deficits noted in some patients can also be associated with an abnormal pulse of innervation to the lateral rectus muscles, which are unable to abduct the eyes due to abducens dysfunction42. Children with GD3 presented with limited abduction, ocular motor apraxia, and esotropia. Therefore, GD should be among the diagnostic hypotheses for patients with these signs43.
Although esotropia can be present in GD, the possibility of it being a consequence of mild abducens palsy cannot be ignored. Hence, it is recommended that patients with GD and esotropia be carefully examined for other eye movement abnormalities before surgery6,18.
The topography of the lesions that cause abnormal eye movements is uncertain, but the movements are believed to be associated with lesions in several places, including the brain stem, cerebellum, basal ganglia, and neocortex41.
DIAGNOSIS
GD is diagnosed by assessing the GCase activity in peripheral blood leukocytes, by fibroblast cell culture on the basis of skin biopsy, or by a molecular analysis of mutations. The residual enzyme activity is usually approximately 10%-15% of the normal level7. Molecular diagnosis can be performed by GBA gene sequencing. The gene that encodes GCase (GBA1) is located on the long arm of chromosome 1 (1q21) and contains 11 exons. The presence of a highly homologous pseudogene (GBAP) on the same location (16 kb downstream) is responsible for recombination events between GBAP and GBA1 (for example, the Rec-NciI allele)44.
The diagnosis of ophthalmological manifestations is performed using eye movement and visual acuity assessment, biomicroscopy of the anterior segment, and full fundoscopic examination. Fluorescein angiography21 and OCT analysis of RNFL31 can also be used.
TREATMENT
The following two different treatment strategies for GD are currently available: intravenous enzyme replacement therapy (ERT) and orally administered substrate reduction therapy (SRT)45.
One of the drugs used in ERT is imiglucerase (Cerezyme®, Sanofi- Genzyme, authorized in 1996), produced using Chinese hamster ovary cells. Other recombinant enzymes were developed later: velaglucerase alfa (Vpriv®, Shire, authorized in 2010), produced using human fibroblasts, and taliglucerase alfa (Elelyso®, Pfizer, authorized in 2011), produced using carrot cells.45
The aim of the other treatment strategy (SRT) is reducing the production of cellular GlcCer (substrate). Miglustat (Zavesca®, Actelion, authorized in 2002) is a GlcCer synthase inhibitor, reducing GlcCer biosynthesis in GC. Although it can cross the blood-brain barrier, miglustat had no significant effect on the neurological symptoms of patients with GD3.7 Another substrate inhibitor is eliglustat (Cerdelga® , Sanofi-Genzyme, authorized in 2015), which does not cross the blood–brain barrier and is only indicated for GD146.
Treatment for ocular manifestations is controversial and lacks evidence of effectiveness, but eye movement has reportedly shown improvement using miglustat (which reduces the synthesis of GlcCer) after two years of treatment40. Posterior segment changes usually do not improve with enzyme replacement medications because of their inability to cross the blood-retinal barrier.21 A few authors reported visual acuity improvement with vitrectomy and epiretinal membrane peeling in patients with GD3.25,47
CONCLUSIONS AND CHALLENGES
Advances in understanding the clinical manifestations of GD are unquestionable; however, the phenotypic heterogeneity of GD for the same gene, the variation in organ involvement, and the complete involvement of macrophages in the pathophysiology of the disease are still to be clarified.
The high cost of GD treatment and the lack of comparative studies of the many treatment strategies challenge researchers. Finding biomarkers that are able to effectively monitor the diagnosis and treatment of GD is one of the aims of several ongoing studies.
Neurological and ophthalmological manifestations are difficult to treat, and drugs capable of effectively controlling these symptoms in patients with GD3 are still being investigated.
Lastly, associations of GD with other diseases are frequently found. In the present review, we present the diversity of ocular findings and highlight the importance of thorough eye examination.
REFERENCES
1. Ginns EI, Choudary PV, Tsuji S, Martin B, Stubblefield B, Sawyer J, et al. Gene mapping and leader polypeptide sequence of human glucocerebrosidase: implications for Gaucher disease. Proc Natl Acad Sci U S A. 1985;82(20):7101-5.
2. Hruska KS, LaMarca ME, Scott CR, Sidransky E. Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum Mutat. 2008;29(5):567-83.
3. Nalysnyk L, Rotella P, Simeone JC, Hamed A, Weinreb N. Gaucher disease epidemiology and natural history: a comprehensive review of the literature. Hematology. 2017;22(2):65-73.
4. Chaves RG, Pereira Lda V, de Araújo FT, Rozenberg R, Carvalho MD, Coelho JC, et al. Consanguinity and founder effect for Gaucher disease mutation G377S in a population from Tabuleiro do Norte, Northeastern Brazil. Clin Genet. 2015;88(4):391-5.
5. Allen MJ, Myer BJ, Khokher AM, Rushton N, Cox TM. Pro-inflammatory cytokines and the pathogenesis of Gaucher’s disease: increased release of interleukin-6 and interleukin-10. QJM. 1997;90(1):19-25.
6. van Breemen MJ, de Fost M, Voerman JS, Laman JD, Boot RG, Maas M, et al. Increased plasma macrophage inflammatory protein (MIP)-1alpha and MIP-1beta levels in type 1 Gaucher disease. Biochim Biophys Acta. 2007;1772(7):788-96.
7. Stirnemann J, Belmatoug N, Camou F, Serratrice C, Froissart R, Caillaud C, et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int J Mol Sci. 2017;18(2):pii: E441.
8. Dunn P, Kuo MC, Sun CF. Pseudo-Gaucher cells in mycobacterial infection: a report of two cases. J Clin Pathol. 2005;58(10):1113-4.
9. Giraldo P, Alfonso P, Irún P, Gort L, Chabás A, Vilageliu L, et al. Mapping the genetic and clinical characteristics of Gaucher disease in the Iberian Peninsula. Orphanet J Rare Dis. 201 Orphanet J Rare Dis. 2012;7:17.
10. Charrow J, Andersson HC, Kaplan P, Kolodny EH, Mistry P, Pastores G, et al. The Gaucher registry: demographics and disease characteristics of 1698 patients with Gaucher disease. Arch Intern Med. 2000;160(18):2835-43.
11. Mistry PK, Sadan S, Yang R, Yee J, Yang M. Consequences of diagnostic delays in type 1 Gaucher disease: the need for greater awareness among hematologists-oncologists and an opportunity for early diagnosis and intervention. Am J Hematol. 2007;82(8):697-701.
12. Pastores GM, Hughes DA. Gaucher Disease. GeneReviews®. Seattle: University of Washington; 1993.
13. Tajima A, Yokoi T, Ariga M, Ito T, Kaneshiro E, Eto Y, et al. Clinical and genetic study of Japanese patients with type 3 Gaucher disease. Mol Genet Metab. 2009;97(4):272-7.
14. Kraoua I, Sedel F, Caillaud C, Froissart R, Stirnemann J, Chaurand G, et al. A French experience of type 3 Gaucher disease: Phenotypic diversity and neurological outcome of 10 patients. Brain Dev. 2011;33(2):131-9.
15. Tylki-Szymańska A, Vellodi A, El-Beshlawy A, Cole JA, Kolodny E. Neuronopathic Gaucher disease: demographic and clinical features of 131 patients enrolled in the International Collaborative Gaucher Group Neurological Outcomes Subregistry. J Inherit Metab Dis. 2010;33(4):339-46.
16. Mejía-Turizo JC, Rojas-Múnera S, Orrego-Betancur SR, Franco-Echeverri CM, Arango-Simoni K. Manifestaciones oculares de la enfermedad de Gaucher: presentación de un caso y revisión del tema. Iatreia. 2017;30(3):297-308.
17. Biswas J, Nandi K, Sridharan S, Ranjan P. Ocular manifestation of storage diseases. Curr Opin Ophthalmol. 2008;19(6):507-11.
18. Guemes A, Kosmorsky GS, Moodie DS, Clark B, Meisler D, Traboulsi EI. Corneal opacities in Gaucher disease. Am J Ophthalmol. 1998;126(6):833-5.
19. East T, Savin LH. A case of gaucher’s disease with biopsy of the typical pingueculae. Br J Ophthalmol. 1940;24(12):611-3.
20. Chu FC, Rodrigues MM, Cogan DG, Barranger JA. The pathology of pingueculae in Gaucher’s disease. Ophthalmic Paediatr Genet. 1984;4(1):7-11.
21. Wang TJ, Chen MS, Shih YF, Hwu WL, Lai MY. Fundus abnormalities in a patient with type I Gaucher’s disease with 12-year follow-up. Am J Ophthalmol. 2005;139(2):359-62.
22. Geens S, Kestelyn P, Claerhout I. Corneal manifestations and in vivo confocal microscopy of Gaucher disease. Cornea. 2013;32(7):e169-72.
23. Petrohelos M, Tricoulis D, Kotsiras I, Vouzoukos A. Ocular manifestations of Gaucher’s disease. Am J Ophthalmol. 1975;80(6):1006-10.
24. Hsing YE, Foster A. Preretinal and posterior vitreous deposits in Gaucher disease. JAMA Ophthalmol. 2014;132(8):992.
25. Shrier EM, Barr CC, Grabowski GA. Vitreous opacities and retinal vascular abnormalities in Gaucher disease. Arch Ophthalmol. 2004;122(9):1395-8.
26. Fujiwaki T, Yamaguchi S, Tasaka M, Takayanagi M, Isobe M, Taketomi T. Evaluation of sphingolipids in vitreous bodies from a patient with Gaucher disease, using delayed extraction matrix-assisted laser desorption ionization time-of-flight mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2004;806(1):47-51.
27. Seidova SF, Kotliar K, Foerger F, Klopfer M, Lanzl I. Functional retinal changes in Gaucher disease. Doc Ophthalmol. 2009;118(2):151-4.
28. Coussa RG, Roos JC, Aroichane M, Miron MC, Ospina LH. Progression of retinal changes in Gaucher disease: a case report. Eye (Lond). 2013;27(11):1331-3.
29. Charters AD. Retinitis pigmentosa and Gaucher’s disease. Br J Ophthalmol. 1957;41(1):54-9.
30. Sheck LH, Wilson CJ, Vincent AL. Analysis of the pre-retinal opacities in Gaucher Disease using spectral domain optical coherent tomography. Ophthalmic Genet. 2012;33(4):253-6.
31. Matos AG, Gurgel VP, Gonçalves MC. Ophthalmologic fndings in Gaucher’s disease type III: case report. Rev Bras Oftalmol. 2017;76(6):316-8.
32. Bruscolini A, Pirraglia MP, Restivo L, Spinucci G, Abbouda A. A branch retinal artery occlusion in a patient with Gaucher disease. Graefes Arch Clin Exp Ophthalmol. 2012;250(3):441-4.
33. Watanabe A, Gekka T, Arai K, Tsuneoka H. A case of traction retinal detachment in a patient with Gaucher disease. Ophthalmic Genet. 2017;38(3):273-6.
34. Chen H, Chan AY, Stone DU, Mandal NA. Beyond the cherry-red spot: Ocular manifestations of sphingolipid-mediated neurodegenerative and inflammatory disorders. Surv Ophthalmol. 2014;59(1):64-76.
35. Dweck A, Rozenman J, Ronen S, Zimran A, Elstein D. Uveitis in Gaucher disease. Am J Ophthalmol. 2005;140(1):146-7.
36. vom Dahl S, Niederau C, Häussinger D. Loss of vision in Gaucher’s disease and its reversal by enzyme-replacement therapy. N Engl J Med. 1998;338(20):1471-2.
37. McNeill A, Roberti G, Lascaratos G, Hughes D, Mehta A, Garway-Heath DF, et al. Retinal thinning in Gaucher disease patients and carriers: results of a pilot study. Mol Genet Metab. 2013;109(2):221-3.
38. Cassidy L, Taylor D, Harris C. Abnormal supranuclear eye movements in the child: a practical guide to examination and interpretation. Surv Ophthalmol. 2000;44(6):479-506.
39. Tsai LP, Sue WC, Hwu WL, Lin KH, Wang TR. Oculomotor apraxia in a case of Gaucher’s disease with homozygous T1448C mutation. Zhonghua Min Guo Xiao Er Ke Yi Xue Hui Za Zhi. 1996;37(1):52-5.
40. Accardo A, Pensiero S, Ciana G, Parentin F, Bembi B. Eye movement impairment recovery in a Gaucher patient treated with miglustat. Neurol Res Int. 2010;2010:358534.
41. Harris CM, Taylor DS, Vellodi A. Ocular motor abnormalities in Gaucher disease. Neuropediatrics. 1999;30(6):289-93.
42. Bremova-Ertl T, Schiffmann R, Patterson MC, Belmatoug N, Billette de Villemeur T, Bardins S, et al. Oculomotor and Vestibular Findings in Gaucher Disease Type 3 and Their Correlation with Neurological Findings. Front Neurol. 2018;8:711.
43. Lee S, Kim HJ, Jeong SY, Hwang JM. Ocular Abnormality of Korean Patients with Molecular Genetically Confirmed Gaucher Disease. J Korean Ophthalmol Soc. 2013;54(1):131-5.
44. Busarla SV, Sadruddin FA, Sohani AR. Pseudo-Gaucher cells in disseminated mycobacterial infection. Am J Hematol. 2013;88(2):155.
45. Zimran A, Szer J. Recent advances and future challenges in Gaucher disease. Blood Cells Mol Dis. 2018;68:9-13.
46. Stone WL, Master SR. Gaucher Disease. Treasure Island: StatPearls Publishing; 2018.
47. Rapizzi E, Vannozzi L, Borgioli V, Giansanti F, Murro V, Sodi A, et al. Vitrectomy for vitreous opacities and macular pucker in Gaucher disease. Eur J Ophthalmol. 2011;21(3):340-2.


Funding: No specifical financial support was available for this study
CEP approval: Not Applicable
Disclosure of potential conflicts of interest: None of the authors have any potential conflito of interest to disclose.
Received on:
April 12, 2018.
Accepted on:
June 7, 2018.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados


 Ler em português
Ler em português
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket