

Marcelo Vicente de Andrade Sobrinho1; Beatriz Crotti Peixoto2; Henrique Sampaio Ferreira2; Giovanna Soares Nutels2
DOI: 10.17545/eOftalmo/2022.0023
RESUMO
Ectopia lentis é o deslocamento da lente (cristalino) de sua posição anatômica normal. A lente pode estar completamente deslocada (luxada) ou parcialmente deslocada (sub-luxada). A ectopia lentis pode ser adquirida ou estar relacionada a doenças oculares e sistêmicas. A fisiopatologia da doença está relacionada à alteração das fibras zonulares, sendo que o deslocamento do cristalino pode causar muitos problemas. Dois casos de lentes de contato associados à ectopia lentis são apresentados, como também uma revisão para discutir as doenças relacionadas a esta síndrome. Documentos da base de dados PubMed foram extraídos usando combinações variáveis dos termos de pesquisa “ectopia lentis”; “lentes de contato”; “síndrome de Marfan”; “Homocistinúria”; “síndrome de Weill-Marchesani”; “trauma ocular”. Artigos relacionados à identificação da ectopia lentis e fatores associados foram incluídos na pesquisa, tais como etiologia, associação sistêmica, diagnóstico e tratamento. As listas de referência dos artigos selecionados foram revistas a fim de se obter artigos adicionais relevantes.
Palavras-chave: Anormalidades congênitas; Lentes de contato; Ectopia lentis; Subluxação do cristalino.
ABSTRACT
Ectopia lentis is the displacement of the lens from its primary position. The lens may be completely (luxated) or partially (subluxated) dislocated. Ectopia lentis can be acquired or related to ocular and systemic diseases. Its physiopathology is related to the disturbance of zonular fibers, and lens displacement can cause many problems. This paper presents two cases of contact lens fitting in ectopia lentis and a review of diseases with this finding. Papers were extracted from the PubMed database using varying combinations of the following search terms: “Ectopia lentis”, “contact lens”, “Marfan syndrome”, “Homocystinuria”, “Weill-Marchesani syndrome”, and “ocular trauma”. Articles related to the identification of ectopia lentis and associated factors, such as etiology, systemic association, diagnosis, and management, were included. The reference lists of the selected articles were reviewed to obtain additional relevant articles.
Keywords: Congenital abnormalities; Contact lenses; Ectopia lentis; Lens subluxation.
INTRODUÇÃO
A ectopia lentis (EL) é o deslocamento da lente (cristalino) de sua posição anatômica normal1-3. A lente pode estar luxada, quando há ruptura das fibras zonulares. Em algumas situações, o cristalino pode migrar para a câmara anterior ou para a câmara vítrea. A lente também pode estar subluxadas, quando há ruptura (ou flacidez) das fibras zonulares, caracterizando uma lente parcialmente deslocada. Neste caso, a lente permanece posicionada na câmara posterior, na zona pupilar3-5.
A EL tem duas origens básicas: hereditária ou secundária a outras causas. Estas últimas incluem trauma, miopia elevada, buftalmia, tumores uveais, síndrome de pseudoexfoliação, cataratas hipermaduras e complicações cirúrgicas2,3,6.
As causas hereditárias podem ser divididas em causas sem associações sistêmicas (por exemplo, ectopia lentis familiar, ectopia lentis et pupillae) e causas com associações sistêmicas (por exemplo, síndrome de Marfan, homocistinúria, síndrome de Weill-Marchesani, hiperlisinemia, deficiência de sulfito oxidase e síndrome de Ehlers Danlos)2,3.
O deslocamento da lente pode levar à anisometropia, diplopia, erros de refração, e ambliopia7-10.
Neste artigo, apresentamos a descrição de dois casos de adaptação de lentes de contato em pacientes com EL, além de uma revisão das doenças relacionadas à EL.
RELATOS DE CASOS
Caso 1
Paciente A.J.C.B., sexo feminino, 6 anos de idade, procedente de Campinas - SP. De acordo com os membros da família, ela tem baixa visão desde o nascimento. Os seus pais são primos de primeiro grau. A paciente não apresentava características sindrômicas. Traumas anteriores ou história familiar de EL foram negados pela família.
Exame oftalmológico:
• Refração: ambos os olhos: +10,00 D.E: Acuidade visual: 20/200
• Biomicroscopia: Ectopia lentis superior e temporal. (Figuras 1 e 2)
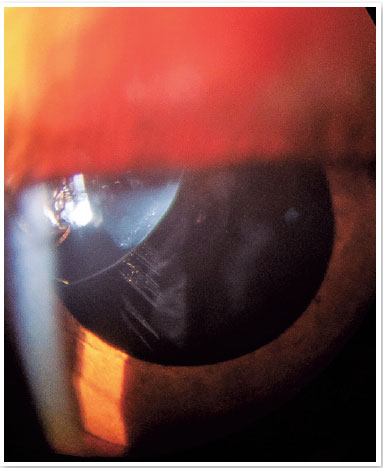
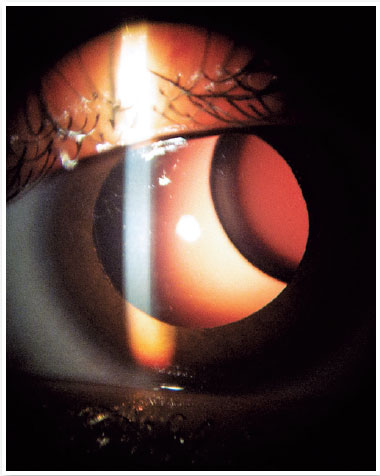
• Fundoscopia: sem alterações.
• Topografia corneana:
OD: 46,23 x 48,76 @ 21°
OS: 46,71 x 49,54 @ 173°
• Conduta: Adaptação de lentes de contato rígidas gás-permeáveis (LCs RGP)
• Parâmetros das LCs:
OD: Curva base (CB): 46,00 D; Poder Dióptrico (PD): +20,00 D; Diâmetro (D) 8,8mm
OS: Curva base (CB): 46,00 D; Poder Dióptrico (PD): +10,00 D; Diâmetro (D) 8,8mm
Lentes de contato esféricas monocurvas
Acuidade visual com lentes de contato: ambos os olhos: 20/100
Caso 2
Paciente A.P.S., sexo masculino, 57 anos de idade, procedente de Campinas - SP. Foi submetido a extração bilateral dos cristalinos após deslocamento espontâneo dos mesmos para a câmara anterior. O paciente não apresentou quaisquer características sindrômicas. Sem relato de evento traumático ou história familiar de EL.
• Refração:
OD: +12,50 -1,50 180 20/30
OE: + 12.00 20/30
• Biomicroscopia:
Ambos os olhos: afacia, corectopia.
• Topografia corneana:
OD: 42,78 x 44,21 @ 148°
OS: 42,92 x 44,05 @ 120°
Conduta: Adaptação de LCs RGP.
• Parâmetros das LCs:
OD: CB: 43.00D; PD: +14.50 D; Diâmetro: 9,6mm (Figura 3)
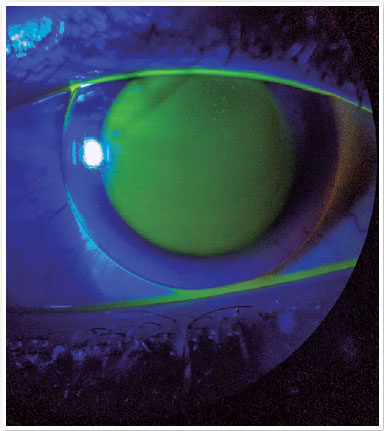
OS: CB: 42.00D; PD: +14.75 D; Diâmetro: 9,6mm
Acuidade Visual com LC:
OD: 20/25
OE: 20/30
Revisão da Literatura
Descrição e etiologia
Berryat foi provavelmente o primeiro a descrever as descobertas clínicas de subluxação do cristalino em um paciente, em 1749. Em 1856, Karl Stellwag von Carion, um oftalmologista tcheco, introduziu o termo “Ectopia Lentis” para um paciente com deslocamento congênito de cristalino. No entanto, foi apenas após muitos anos que a EL foi associada a outras doenças oculares e sistêmicas2.
Na Ectopia Lentis, os sinais e sintomas dependem da extensão da subluxação11. A baixa de acuidade visual é o principal sintoma. O grau de deslocamento da lente, associado a outras alterações oculares, pode levar a ametropias e/ou anisometropias, além da baixa acuidade visual7,12.
O deslocamento anterior e as rotações em torno do eixo da lente podem levar a miopia lenticular. Isto pode acontecer devido ao relaxamento das fibras zonulares2,13. Se a borda da lente estiver em frente à zona pupilar, o paciente pode perceber distorções ópticas e diplopia. Quando isto ocorre de forma bilateral, gera-se quadriplopia2,3.
A EL está associada a numerosas complicações que pioram o prognóstico visual do paciente, tais como catarata, glaucoma secundário (devido à malformação do ângulo da câmara anterior) e descolamento da retina10,12,14.
O sistema de classificação de Ectopia Lentis (C.E.L.) classifica a EL de acordo com o movimento da lente (deslocamento ou subluxação), sendo o deslocamento definido como movimento completo, que pode ser anterior ou posterior, e a subluxação definida como movimento da lente no plano coronal atrás da íris. Este sistema de classificação é relevante para a padronização da avaliação e do detalhamento do movimento da lente2,3.
EL sem associações sistêmicas
EL simples ou EL familiar
A EL simples, ou EL familiar, é uma doença autossômica dominante sem manifestações sistêmicas associadas. A existência de uma forma autossômica recessiva foi também relatada15-18.
Pode apresentar-se como uma condição clínica congênita ou espontânea com início tardio, sendo esta última a forma mais comum, ocorrendo entre os 20 e 65 anos de idade2,19.
As mutações ocorrem no gene fibrilina-1 (FBN1), mapeado para o cromossoma 15q213.
Pode haver uma sobreposição entre a genética da EL simples e doenças sistêmicas associadas à EL, uma vez que, as mutações no gene FBN1 também podem ocorrer na síndrome de Marfan9,20,21.
Nesta condição, há irregularidade e degeneração das fibras zonulares, causando subluxação da lente. A sua manifestação principal é o deslocamento bilateral, simétrico, superior e temporal da lente5,17,20. Contudo, em alguns casos, o grau de deslocamento da lente pode variar entre os dois olhos e ser assimétrico2,3. Possíveis complicações são: descolamento da retina, glaucoma secundário e deslocamento completo da lente20,22. Estes pacientes podem desenvolver alterações cardiovasculares e devem ter um acompanhamento cardiológico a longo prazo devido à relação com o gene da síndrome de Marfan20,23.
EL et pupillae
EL et pupillae uma doença congênita rara e é caracterizada pela ectopia concomitante da pupila e da lente10.
Trata-se de uma doença autossômica recessiva, e pode estar relacionada com uma mutação no gene ADAMTSL4, no cromossomo 1q2124-26.
O diagnóstico é essencialmente clínico e importante para a avaliação de risco, prognóstico e tratamento10,27. Na maioria dos casos, há um envolvimento de ambos os olhos, que pode ser simétrico ou não, sendo as pupilas retraídas, ovais ou elípticas e deslocadas na direção oposta à da EL28,29. No entanto, 40% apresentam apenas uma dilatação pupilar discreta18,28,30. Anormalidades oculares, síndromes e desordens metabólicas fazem parte do diagnóstico diferencial desta doença24,27. Sua patogênese ainda é desconhecida. Entretanto, há teorias que advogam a ocorrência de alterações mesodérmicas e neuroectodérmicas17,26,31. Com estas alterações, há a formação de um tecido vascularizado persistente e anormal que se anastomosa com o sistema hialoide, gerando ectopia pupilar e malformação da zônula na área correspondente10,17,26.
Casos com microesferofacia e transiluminação da íris também foram documentados na literatura10,25,26.
A uveíte é relativamente comum e os principais mecanismos são o contato direto do cristalino deslocado com o corpo ciliar ou com a face posterior da íris, levando à iridociclite aguda, e ao aumento da permeabilidade ou ruptura da cápsula anterior do cristalino, com escape de proteínas, causando a uveíte facolítica24,30.
EL com associações sistêmicas
Síndrome de Marfan
Esta síndrome foi descrita em 1896 pelo pediatra francês Bernard-Jean Antoine Marfan. É uma desordem autossômica dominante do tecido conjuntivo causada por uma mutação no gene FBN1 no cromossoma 15q212,3.
As manifestações incluem envolvimento cardiovascular, ocular e esquelético. A doença não tem predileção por gênero, afetando tanto homens como mulheres igualmente32,36,38.
Os achados clínicos esqueléticos incluem crescimento excessivo de ossos longos, gerando deformidade torácica, membros desproporcionais, escoliose, aracnodactilia, entre outros36,37. Mialgia, fadiga e hipoplasia muscular estão normalmente presentes2,9.
Os pacientes com envolvimento cardiovascular apresentam disfunção da válvula mitral, com prolapso, insuficiência e calcificação. As manifestações cardíacas são a principal causa de morbidade e mortalidade da síndrome de Marfan. Dilatação da aorta, dissecção, aneurisma e até ruptura da raiz aórtica podem ocorrer9,38,39.
EL é a manifestação ocular mais comum, e afeta cerca de 80% dos pacientes, sendo que 70% dos casos ocorrem antes dos 6 anos de idade3,9. A subluxação ocorre principalmente na região temporal superior ou na região nasal superior, e há irregularidade na borda da lente; as fibras zonulares são longas e podem sofrer rupturas ou retração completamente14,22,32. O coloboma de cristalino também pode ocorrer, mas é uma manifestação ocular menos comum. Caracteriza-se por um entalhe no equador da lente, com zônulas ausentes ou subdesenvolvidas, sendo geralmente monocular e pode ou não estar associado a outras anormalidades sistêmicas34. Outros achados oftalmológicos podem incluir exoftalmia (devido à diminuição do tecido adiposo retro-orbital), megalocórnea, transiluminação da íris, miopia alta, descolamento da retina, catarata e glaucoma40,41.
Existe uma classificação de EL na síndrome de Marfan, que avalia o valor preditivo positivo da EL de acordo com o exame oftalmológico, dividida em 5 fases: fase 1 (deslocamento antero-posterior - deslocamento mínimo), fase 2 (deslocamento antero-posterior e deslocamento superior), fase 3 (subluxação e alongamento das fibras zonulares inferiores), fase 4 (subluxação e ruptura de algumas fibras zonulares), fase 5 (deslocamento). A partir da fase 2 há um elevado valor preditivo positivo para EL e diagnóstico clínico de Síndrome de Marfan37.
Em 2010, os critérios nosológicos de Ghent, utilizados para o diagnóstico da síndrome de Marfan, foram atualizados33.
O aumento da expectativa de vida na síndrome de Marfan está relacionado à abordagem precoce das complicações cardiovasculares39.
Homocistinúria
Essa doença é caracterizada pela deficiência das enzimas cistationina β-sintase e metionina sintase, responsáveis pelo metabolismo da homocisteína, e é causado por mutações no gene CBS, no cromossomo 21q2233. A sua herança é autossômica recessiva42.
A homocisteína é detectada em testes de urina, mas pode estar presente em condições além da homocistinúria. A cromatografia de urina ou eletroforese de alta voltagem são exames que confirmam o diagnóstico2.
O acúmulo de homocisteína irá gerar manifestações cardiovasculares, esqueléticas e oculares. As principais manifestações são atraso cognitivo progressivo e subluxação inferonasal do cristalino (está presente em cerca de 90% dos pacientes, é progressiva e ocorre devido a alterações degenerativas nas fibras zonulares)42,43.
A miopia lenticular também é comum e geralmente precede o surgimento da EL2,14. Esses pacientes têm um aspecto semelhante ao dos pacientes com síndrome de Marfan e podem ter eventos tromboembólicos precoces e anormalidades esqueléticas, incluindo osteoporose, geno valgo, desbaste e alongamento dos ossos longos. A principal causa de mortalidade da síndrome está relacionada com oclusões vasculares trombóticas42.
O paciente deve manter uma dieta pobre em metionina e rica em cisteína com suplemento de piridoxina (vitamina B6), o que pode prevenir ou atrasar o retardo mental e a subluxação do cristalino11,43.
Síndrome de Weill-Marchesani
Esta síndrome foi descrita por Georges Weill em 1932 e delineada por Oswald Marchesani sete anos mais tarde3.
Trata-se de uma síndrome rara causada por uma mutação genética nas proteínas ADAMTS ou FBN1, que gera uma desordem do tecido conjuntivo e pode ser de herança autossômica dominante ou recessiva, com braquimorfismo ocasional nos heterozigotos2,3,44.
A síndrome caracteriza-se clinicamente pela microesferofacia (em alguns casos, a lente tem 50% do seu volume normal), baixa estatura, braquidactilia, pele espessa, articulações rígidas e defeitos cardíacos3. A osteoporose também tem sido descrita nesta síndrome45.
A microesferofacia é uma manifestação ocular primária, e é considerada um critério de diagnóstico para esta síndrome. Caracteriza-se por um cristalino pequeno, esférica, com fibras zonulares menores, alongadas e relaxadas em torno do seu equador13. A miopia lenticular e o deslocamento inferior da lente também podem estar presentes. Há também uma elevada prevalência de glaucoma associado devido ao deslocamento da lente e ao contato com a íris, o que pode causar perda visual3,44. Outras anomalias oculares associadas à síndrome de Weill-Marchesani, são megalocórnea, degeneração coriorretiniana e estafiloma escleral2,3.
Hiperlisinemia
É uma doença sistêmica rara, caracterizada pelo aumento anormal da lisina, um aminoácido essencial. A hiperlisinemia é uma doença autossômica recessiva e ocorre devido à mutação do gene AASS, no cromossomo 7q31.3s. Tem alta associação com a consanguinidade2,3.
Clinicamente, os pacientes têm atraso cognitivo e hipotonia muscular. Podem também apresentar EL, vômitos recorrentes, letargia, diarreia e atraso do desenvolvimento3,19.
Quando a EL ocorre, apresenta-se como subluxação bilateral e pode estar associada à paresia dos músculos extra-oculares2.
Deficiência de sulfito oxidase
O primeiro caso desta síndrome foi descrito em 1967 por Irrevere. É uma doença rara, autossômica dominante, e o gene envolvido é o gene SUOX (Human sulfite oxidase) que codifica a enzima sulfito oxidase humana, localizado no cromossomo 12q13.13. Há uma alteração no metabolismo da metionina e da cisteína, o que gera um aumento da excreção urinária de S-sulfo-cisteína, taurina, sulfito e tiossulfato2,3.
Nesta síndrome, há envolvimento do sistema nervoso central devido ao acúmulo de sulfito no cérebro, gerando perdas graves e generalizadas de neurônios, mielina e axônios, com proliferação de células gliais. Portanto, o paciente pode apresentar alterações no tônus muscular, convulsões refratárias, distonia e atraso cognitivo2,19.
A EL foi relatada em 53% dos casos, sendo diagnosticada entre 3 meses e 3 anos de idade. A patogênese da EL na deficiência de sulfito oxidase ainda não é clara3.
Síndrome de Ehlers-Danlos
Esta síndrome leva os nomes dos dermatologistas Edvard Ehlers, dinamarquês, e Henri-Alexandre Danlos, francês, que a descreveram, respectivamente, em 1901 e 190846.
É uma doença rara, causada, em 50% dos seus tipos clássicos, por mutações nos genes COL5A1 (cromossoma 9q34.3) e COL5A2 (cromossoma 2q32.2), os quais codificam as cadeias alfa1 e alfa2 do colágeno tipo V. Não há predisposição racial e afeta cerca de 1 em cada 5.000 nascidos vivos3.
Caracteriza-se pela fragilidade generalizada dos tecidos conjuntivos moles, pela hiperextensibilidade cutânea, atraso de cicatrização, hipermobilidade articular e hematomas. A EL ocorre ocasionalmente, e outras manifestações oculares podem estar presentes, tais como fragilidade escleral, ceratocone e miopia3.
Diagnóstico
Ao atender um paciente com EL, o médico deve prestar atenção à idade e à história pessoal e familiar de comorbidades do paciente. O exame físico deve incluir busca por características sindrômicas, alterações esqueléticas, cardiovasculares e oculares14,30
Quando o paciente se encontra em midríase, às vezes é possível se observar a borda do cristalino e as fibras zonulares.
Todo paciente com suspeita de EL hereditária deve ser submetido a avaliação sistêmica com rastreio metabólico, ecocardiografia e exame músculo-esquelético7,19,39.
A gonioscopia deve ser realizada em todos os pacientes, pois pode ocorrer estreitamento angular. Alterações iridocorneanas, tais como megalocórnea e corectopia, também devem ser investigadas27,30.
Exames de imagem podem ser utilizados para auxílio diagnóstico, tais como ultrassonografia ocular, tomografia computadorizada e ressonância magnética. A ultrassonografia é a primeira opção em termos de disponibilidade, e é capaz de identificar complicações como descolamento da retina47.
Conduta
O acompanhamento clínico do paciente e a correção óptica com óculos ou lentes de contato continuam a ser a abordagem clássica para a EL, especialmente quando a subluxação é pequena, estável e não há complicações9,48-50.
A prescrição de correção óptica é útil para a prevenção de ambliopia, especialmente em pacientes com erros de refração baixos e estáveis14,30.
As lentes de contato têm várias vantagens sobre os óculos, tais como menos aberrações ópticas, eliminação de distorções periféricas e de efeitos prismáticos. As lentes rígidas gás-permeáveis são a melhor opção em termos de acuidade visual, mas alguns autores recomendam que as lentes de contato feitas de silicone sejam a primeira escolha porque têm maior permeabilidade ao oxigênio, condutividade térmica, facilidade de colocação e relativa segurança. Assim, a escolha do tipo de lente depende de questões adaptativas da criança e da cooperação da família51.
O tratamento cirúrgico pode ser necessário quando há presença de astigmatismo lenticular elevado, rotação da lente no seu próprio eixo, mobilidade da lente com refrações instáveis e presença da borda da lente no eixo visual52,53. Nestes pacientes, cada caso deve ser avaliado individualmente, uma vez que existem riscos elevados inerentes aos procedimentos cirúrgicos. De modo geral, quanto menor for o grau de subluxação, mais seguro é o procedimento cirúrgico54-56.
A escolha da técnica cirúrgica depende do paciente, das condições oculares, da doença que causou o deslocamento da lente e da habilidade, experiência e preferência do cirurgião. Embora o procedimento cirúrgico ainda apresente resultados limitados, a margem de sucesso do tratamento tem aumentado, uma vez que, tem ocorrido um grande avanço das técnicas e materiais cirúrgicos55,56.
Algumas das técnicas cirúrgicas mais utilizadas são a facectomia extracapsular, a facoemulsificação com ou sem implantação de lente intraocular (LIO) no sulco ciliar ou saco capsular, e a facofragmentação com vitrectomia posterior via pars plana com fixação de LIO na esclera ou na íris. Durante a cirurgia, deve ser utilizado um anel expansor endocapsular para melhor visualização, especialmente na facectomia extracapsular ou na facoemulsificação52,57.
A indicação cirúrgica para EL mais comum é quando a borda do cristalino atinge o eixo visual, quando há desinserção extensa da zónula, ou quando há falta de suporte capsular50,58. Nestes casos, a lensectomia-vitrectomia é realizada através da pars plana ou por via limbal. A afacia deve ser corrigida com lentes de contato, óculos ou implante de LIO para evitar a ambliopia59,60. Quando se escolhe a correção com implante de LIO, é necessário um acompanhamento de longo prazo devido ao risco de ruptura da sutura e consequente deslocamento da lente intraocular ao longo do tempo. A fixação escleral da LIO na câmara posterior é o procedimento de escolha atual na ausência de suporte capsular adequado59-62.
Quando a lente tem um deslocamento de mais de 180°, a utilização do anel capsular de Cionni permite a melhor visualização cirúrgica e fixação do saco capsular na esclera. O anel também mantém a integridade do saco capsular em lesões zonulares extensas, evitando o colapso do saco capsular após a remoção do cristalino, além de oferecer segurança durante a facoemulsificação e implante da LIO, mantendo a cápsula estável63-66.
Na EL associada à síndrome de Marfan, se o paciente não obtiver boa acuidade visual com correção óptica, ou apresentar complicações, a lensectomia com ou sem implante secundária de LIO é indicada40,53,65.
Na EL et pupillae, a principal abordagem é clínica. A correção óptica precoce deve ser realizada para evitar a ambliopia. Em pacientes que têm pupila fora do eixo, a iridectomia deve ser realizada10,12.
Em EL decorrente de homocistinúria, o tratamento cirúrgico deve ser considerado, especialmente em casos de deslocamento da lente para a câmara anterior ou glaucoma por bloqueio pupilar67.
O deslocamento espontâneo da lente para a câmara anterior, como ocorreu em um dos casos descritos nesse artigo, é considerado uma urgência oftalmológica, devido ao risco de descompensação da córnea e de glaucoma68. Complicações pós-operatórias, tais como perda de vítreo e descolamento da retina, podem ser evitadas com lensectomia e vitrectomia via pars plana7,11.
O aconselhamento genético deve ser sempre considerado. Os pacientes devem evitar esportes de contato, a fim de evitar traumas, ou manobras que possam causar um aumento da pressão intraocular, uma vez que as complicações oculares são mais frequentes nestes pacientes do que na população normal14.
Atualmente, o tratamento clínico é a primeira opção, uma vez que, o tratamento cirúrgico pode apresentar várias complicações.
No primeiro caso apresentado neste artigo, se considerarmos a idade do paciente, a cirurgia exporia a criança a uma série de complicações, tais como afacia, uveíte, sinéquias anteriores e posteriores, glaucoma secundário, descolamento da retina, entre outras. A utilização de LC é uma alternativa para adiar a cirurgia até a criança atingir uma idade em que as taxas de complicações são mais baixas. As lentes de contato são capazes de proporcionar uma acuidade visual satisfatória para desenvolver a visão e prevenir ambliopia.
No segundo caso, a adaptação da LC também foi útil, pois proporcionou uma boa acuidade visual e deu ao paciente uma estética melhor.
O gerenciamento da EL continua a ser um desafio, especialmente em crianças.
REFERÊNCIAS
1. AlShehri OA, Almarzouki H, Alharbi BA, Alqahtani M, Allam K. Deslocamentos bilaterais posteriores de lentes cristalinas numa criança de outra forma saudável. Casos de GMS Ophthalmol. 2017 Oct 20;7:Doc26.
2. Chandra A, Patel D, Aragon-Martin JA, Pinard A, Collod-Béroud G, Comeglio P, et al. A nosologia ghent revista; reclassificando ectopia lentis isolada. Clin Genet. 2015;87(3):284-7.
3. Parapia LA, síndrome de Jackson C. Ehlers-Danlos - uma revisão histórica. Br J Haematol. 2008;141(1):32-5.
4. Nelson LB, Maumenee IH. Ectopia lentis. Surv Ophthalmol. 1982;27(3):143-60.
5. Baikoff G. Aspiração de Ectopia Lentis. Dev Ophthalmol. 1985; 11:157-61.
6. Tartarella MB, Araújo Filho A, Sallum JMF, Erwenne CM. Ectopia Lentis et Pupillae. Arq Bras Oftalmol. 1994;57(1):30-3.
7. Severo NS, Kleinert F, Kwitko S. Abordagem cirúrgica da lente subluxada. Arq Bras Oftalmol. 2004;67(1):9-12.
8. Saatci AO, Soylev M, Kavukçu S, Durak I, Saatci I, Memisoglu B. Megalocornea bilateral com subluxação unilateral da lente. Genet oftálmico. 1997;18(1):35-8.
9. Yen KG, Reddy AK, Weikert MP, Song Y, Hamill MB. Lentes intra-oculares de câmara posterior com íris em crianças. Am J Ophthalmol. 2009;147(1):121-6.
10. Oliveira DF, Marchi PH, Arieta CEL. Resultado visual após lensectomia para lentes sublimadas em crianças. Medicina (Ribeirão Preto). 2002;35:62-9.
11. Adès LC, Holman KJ, Brett MS, Edwards MJ, Bennetts B. Ectopia lentis fenotypes e o gene FBN1. Am J Med Genet A. 2004;126A(3):284-9.
12. Packer M, Fine IH, Hoffman RS. Fixação de sutura de uma lente intra-ocular em acrílico dobrável para ectopia lentis. J Catarata Refract Surgimento. 2002;28(1):182-5.
13. Marcio F, Ramos GZ, Moreira PB, Thiesen EB, Souza LB. Ectopia lentis et pupillae. Rev Bras Oftalmol. 2011;70(3):182-4.
14. Booms P, Withers AP, Boxer M, Kaufmann UC, Hagemeier C, Vetter U, et al. Uma novela de nova mutação no exão 14 do gene da fibrilina-1 associada à secreção retardada de fibrilina num paciente com um fenótipo de Marfan ligeiro. Hum Genet. 1997;100(2):195-200.
15. Ekonomidis P, Androudi S, Brazitikos P, Alexandridis A. Ectopia lentis et pupillae: relatório de um caso unilateral e gestão cirúrgica. Graefes Arch Clin Exp Ophthalmol. 2006;244(7):878-9.
16. Varga B. Os resultados das minhas operações melhorando a acuidade visual da ectopia lentis. Ophthalmologica. 1971;162(2): 98-110.
17. Byles DB, Nischal KK, Cheng H. Ectopia lentis et pupillae. Uma hipótese revisitada. Oftalmologia. 1998;105(7):1331-6.
18. Sabrane I, Saoudi S, Ikhloufi ME, Elkaissoumi L, Taouri N, Amazouzi A, et al. Ectopia lentis na homocistinúria. J Fr Ophtalmol. 2019; 42(2):219-20.
19. Meyer ET. A ectopia lentis familiar e as suas complicações. Br J Ophthalmol. 1954;38(3):163-72.
20. Sharifi Y, Tjon-Fo-Sang MJ, Cruysberg JRM, Maat-Kievit, AJ. Ectopia lentis et pupillae em quatro gerações causadas por novas mutações no gene ADAMTSL4. Br J Ophthalmol. 2013;97(5):583-7.
21. Salame AL, Simon EJ, Leal F, Lipener C, Brocchetto D. Lente de contacto em crianças: aspectos epidemiológicos. Arq Bras Oftalmol. 2008;71(3):348-51.
22. Vasavada AR, Praveen MR, Desai C. Gestão da deslocação anterior bilateral de uma lente numa criança com síndrome de Marfan. J Catarata Refract Surgimento. 2003;29(3):609-13.
23. Rødahl E, Mellgren AEC, Boonstra NE, Knappskog PM. ADAMTSL4-Related Eye Disorders. 2012. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, Amemiya A, editores. GeneReviews® [Internet]. Seattle (WA): Universidade de Washington, Seattle; 1993-2021.
24. Chandra A, Charteris D. Patogénese Molecular e estratégias de gestão da ectopia lentis. Olho (Lond). 2014;28(2):162-8.
25. Fuchs J. Marfan e outras doenças sistémicas com ectopia lentis congénita. Um inquérito nacional dinamarquês. Acta Paediatr. 1997;86(9):947-52.
26. Luebbers JA, Goldberg MF, Herbst R, Hattenhauer J, Maumenee AE. Transiluminação Iris e expressão variável em ectopia lentis et pupillae. Am J Ophthalmol. 1977;83(5):647-56.
27. Bernardes CS, Leite LVO, Castro FAA. Ectopia lentis et pupillae: Relato de Caso. Arq Bras Oftalmol. 2005;68(6):841-4.
28. Cline JW, Goyer RA, Lipton J, Mason RG. Homocistinúria adulta com ectopia lentis. Sul Med J. 1971;64(5):613-7.
29. Thapa BB, Agarwal A, Singh R, Gupta PC, Ram J. Phacoaspiration com um anel Cionni versus pars plana lensectomia, vitrectomia e fixação trans-cleral sem sutura de IOL em pacientes pediátricos com uma lente subluxada. Graefes Arch Clin Exp Ophthalmol. 2016;254(5):901-9.
30. Goldberg MF. Manifestações clínicas de ectopia lentis et pupillae em 16 pacientes. Oftalmologia. 1988;95(8):1080-7.
31. Neiva AG, Cunha RNP, Ferreira RC, Erwenne CM. Causas de cristalino ectópico em um hospital universitário. Arq Bras Oftalmol. 1995;58(5):307-9.
32. Fuchs J, Rosenberg T. Congenital ectopia lentis. Um inquérito nacional dinamarquês. Escândalo Acta Ophthalmol. 1998;76(1): 20-6.
33. Chandra A, Banerjee PJ, DG Charteris. Classificação em ectopia lentis (GEL): um novo sistema de classificação. Br J Ophthalmol. 2013;97(7):942-3.
34. Halpert M, BenEzra D. Cirurgia da lente subluxada hereditária em crianças. Oftalmologia. 1996;103(4):681-6.
35. Kopel AC, Carvounis PE, Hamill MB, Weikert MP, Holz ER. Lentes intra-oculares com sutura de íris para ectopia lentis em crianças. J Catarata Refract Surgimento. 2008;34(4):596-600.
36. Sallum JMF, Chen J, Perez ABA. Anomalias oculares e características genéticas na síndrome de Marfan. Arq Bras Oftalmol. 2002;65(6):623-28.
37. Sahay P, Shaji KR, Maharana PK, Titiyal JS. Deslocação anterior espontânea da lente num caso de ectopia lentis et pupillae: uma entidade rara tratada por uma nova técnica de tomografia de coerência óptica integrada ao microscópio (MIOCT) guiada por aspiração intralenticular da lente. BMJ Case Rep. 2019;12(1):bcr-2018-227047.
38. Sadiq MA, Vanderveen D. Genetics of ectopia lentis. Semin Ophthalmol. 2013;28(5-6):313-20.
39. Eken C, Yuruktumen A, Yildiz G. Diagnóstico ultra-sonográfico de luxação traumática da lente. J Med. Emerg. 2013;44(1):e109-10.
40. Zech JC, Putoux A, Decullier E, Fargeton AE, Edery P, Plauchu H, et al. Classificação da Ectopia Lentis na Síndrome de Marfan em Cinco Graus de Grau de Gravidade Crescente. J Clin Med. 2020;9(3):721.
41. McGavic JS. Síndrome de Weill-Marchesani. Brachymorphism e ectopia lentis. Am J Ophthalmol. 1966;62(5):820-3.
42. Neely DE, Plager DA. Gestão da ectopia lentis em crianças. Ophthalmol Clin North Am. 2001;14(3):493-9.
43. Giordano N, Senesi M, Battisti E, Mattii G, Gennari C. Síndrome de Weill-Marchesani: relato de um caso invulgar. Tecido Calcif Tissue Int. 1997;60(4):358-60.
44. Rossiter JD, Morris AH, Etchells DE, Crick MP. Vitrectomia para glaucoma facolítico num paciente com ectopia lentis et pupillae. Olho (Lond). 2003;17(2):243-4.
45. Cruysberg JR, Pinckers A. Ectopia lentis et pupillae syndrome em três gerações. Br J Ophthalmol. 1995;79(2):135-8.
46. Rezar-Dreindl S, Stifter E, Neumayer T, Papp A, Gschliesser A, Schmidt-Erfurth U. Resultado visual e resultados cirúrgicos em crianças com síndrome de Marfan. Clin Exp Ophthalmol. 2019;47(9):1138-45.
47. Farnsworth PN, Burke PA, Blanco J, Maltzman B. Anormalidades ultra-estruturais numa lente ectópica microesférica. Res. Olhos Exp. 1978;27(4):399-408.
48. Ruiz C, Rivas F, Villar-Calvo VM, Serrano-Lucas JI, Cantu JM. Ectopia lentis simples familiar. Uma provável forma autossómica recessiva. Ophthalmic Paediatr Genetatr. 1986;7(2):81-4.
49. Waiswol M, Abujamra S, Cohen R, Almeida GV. Variação da acuidade visual em pacientes jovens com ectopia lentis submetidos à cirurgia. Arq Bras Oftalmol. 2005;68(4):495-504.
50. Arraes C, Endriss D, Lobato F, Arraes J, Ventura M. Subluxação congênita do cristalino: resultados visuais e posição das lentes intraoculares após uma cirurgia. Arq Bras Oftalmol. 2010;73(2):171-4.
51. Seetner AA, Crawford JS. Correcção cirúrgica da luxação da lente em crianças. Am J Ophthalmol. 1981;91(1):106-10.
52. Hoffman RS, Snyder ME, Devgan U, Allen QB, Yeoh R, Braga-Mele R, ASCRS Comité Clínico da Catarata; Subcomité de Cirurgia da Catarata Desafiante/Complicada. Gestão da lente cristalina subluxada. J Catarata Refract Surgimento. 2013;39(12):1904-15.
53. Ventura M, Endriss D. Implantação de lente intraocular com uma alça amputada: proposta para o tratamento cirúrgico da subluxação do cristalino. Arq Bras Oftalmol. 2010;73(2):135-40.
54. Aandekerk AL, Cruysberg JRM. Fotografia de ectopia lentis. J Audiov Media Med. 1987;10(3):87-9.
55. Jaureguy BM, Hall JG. Ectopia lentis congénita isolada com herança autossómica dominante. Clin Genet. 1979;15(1):97-109.
56. Hsu HY, Edelstein SL, Lind JT. Gestão cirúrgica da ectopia lentis pediátrica não-traumática: Uma série de casos e revisão da literatura. Saudi J Ophthalmol. 2012;26(3):315-21.
57. Harrison DA, Mullaney PB, Mesfer SA, Awad AH, Dhindsa H. Gestão das complicações oftálmicas da homocistinúria. Oftalmologia. 1998;105(10):1886-90.
58. Sahu S, Yadav R, Gupta S, Raj Puri L. Ectopia lentis bilateral com coloboma de lente isolada na síndrome de Marfan. Casos de GMS Ophthalmol. 2016 Dec 5;6:Doc14.
59. Sen P, Attiku Y, Bhende P, Rishi E, Ratra D, Sreelakshmi K. Resultado da sutura de lentes intra-oculares fixadas por sutura na síndrome de Marfan em olhos pediátricos. Int Ophthalmol. 2020;40(6):1531-8.
60.Tsai WS, Lee YC, Chang FL, He MS. Lentes decentes duplas num olho: um dilema terapêutico na síndrome de Marfan. Clin Exp Optom. 2020;103(6):911-2.
61. Buchta RM. Ectopia lentis et pupillae. Clin Pediatr (Phila). 1974;13(12):1079-80.
62. Olgun DÇ, Kantarci F. Imagens em medicina clínica: Ectopia lentis. N Engl J Med. 2015;372(9):e13.
63. Simon MA, Origlieri CA, Dinallo AM, Forbes BJ, Wagner RS, Guo S. Novas Estratégias de Gestão para a Ectopia Lentis. J Pediatr Ophthalmol Strabismus. 2015;52(5):269-81.
64. Wu-Chen WY, Letson RD, Summers CG. Resultados funcionais e estruturais após lensectomia para ectopia lentis. J AAPOS. 2005;9(4):353-7.
65. Nb K, Kohli P, Pangtey BPS, Ramasamy K. Avaliação da Lente Intraocular Sem Sutura, Sem Cola, Sem Ectopia Lentis, Sem Ectopia Lentis, Sem Ectopia Lentis. J Ophthalmol. 2018 Ago 29;2018:3212740.
66. Konradsen T, Kugelberg M, Zetterström C. Resultados visuais e complicações na cirurgia da ectopia lentis em crianças. J Catarata Refract Surgimento. 2007;33(5):819-24.
67. al-Salem M. Autosomal recessivo ectopia lentis em dois pedigrees da família árabe. Ophthalmic Paediatr Genetatr. 1990;11(2):123-7.
68. Waiswol M, Kasahara N. Sistema de classificação de subluxação de lentes: valor preditivo para resultados cirúrgicos da ectopia lentis. Einstein (São Paulo). 2009;7(1):81-7.
INFORMAÇÃO DOS AUTORES




Financiamento: Declaram não haver
Conflitos de Interesse: Declaram não haver
Recebido em:
24 de Junho de 2021.
Aceito em:
1 de Junho de 2022.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados
 Ler em inglês
Ler em inglês
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket