

Felipe Leão de Lima; Charles Porto Petruceli Carayon; Lara Lopes de Almeida
DOI: 10.17545/eOftalmo/2021.0032
RESUMO
A neurofibromatose é uma doença autossômica dominante com apresentação clínica variada acometendo diversos tecidos e órgãos, podendo se apresentar com rica manifestação oftalmológica. O presente trabalho apresenta o caso clínico de uma criança, 8 anos de idade, diagnosticada com neurofibromatose tipo 1 em exame oftalmológico eletivo.
Palavras-chave: neurofibromatose; Von Recklinghausen; Nódulos de Lisch; NF1; Manchas café-com-leite; Glioma; nervo óptico.
ABSTRACT
Neurofibromatosis is an autosomal dominant disease with varying clinical presentations affecting several tissues and organs and may present with remarkable ophthalmological manifestation. This study presents the clinical case of an 8-year-old child diagnosed with type 1 neurofibromatosis during an elective ophthalmologic examination.
Keywords: Neurofibromatosis; Von Recklinghausen; Lisch nodules; NF1; café au lait macules; Glioma; Optic nerve.
INTRODUÇÃO
A neurofibromatose representa um espectro de doenças de origem autossômica dominante com incidência aproximada de 1/3.000 nascidos vivos1, caracterizada por síntese insuficiente da proteína neurofibromina - uma proteína supressora de tumores - e consequente desordem de neurônios, oligodendócitos, astrócitos, leucócitos e medula das suprarrenais2.
Possui diagnóstico essencialmente clínico, podendo ser comprovado através de biópsia das tumorações3. O conhecimento desse grupo de patologias se faz importante na prática clínica do oftalmologista, sendo ele muitas das vezes o responsável pelo diagnóstico inicial e condução precoce da condição. Esse é o relato de um caso de NF1 associado a glioma de nervo óptico com diagnóstico realizado através de alterações importantes do exame oftalmológico.
RELATO DE CASO
D.M.C, 8 anos, submetido a consulta oftalmológica eletiva por queixa de baixa de acuidade visual preferencialmente à esquerda, de início insidioso e progressivo há aproximadamente 6 meses.
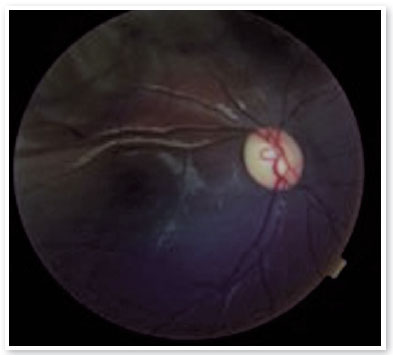
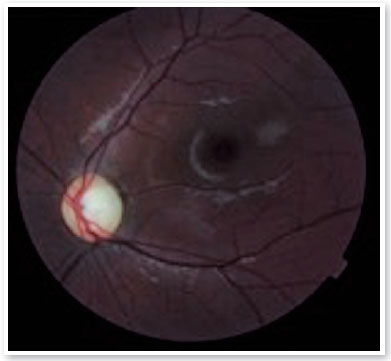
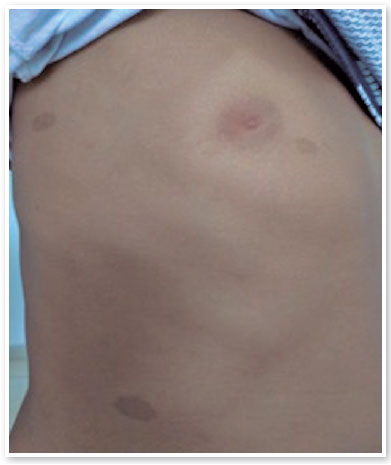
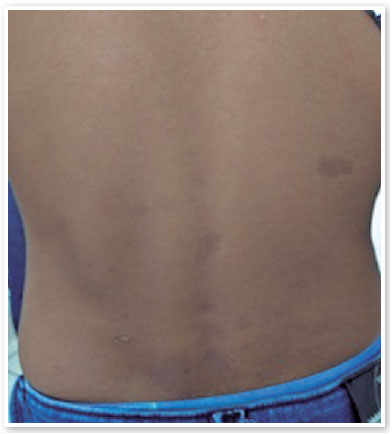
Ao exame oftalmológico, refração estática de +1,50D esférica em ambos os olhos (AO) com acuidade visual de 20/20 em olho direito (OD), 20/100 parcial em olho esquerdo (OE) e alteração no teste de cores de Ishihara. Motilidade ocular extrínseca sem alterações. À biomicroscopia, pupilas isocóricas e fotorreativas com nodulações hamartomatosas irianas em AO, mais concentradas inferiormente, sugestivas de nódulos de Lisch (Figura 1). Gonioscopia demonstrando ângulo aberto até corpo ciliar em AO. Fundoscopia demonstrou palidez de papila óptica bilateral mais acentuada em OE, relação escavação/disco de 0,4 OD e 0,8 OE (Figuras 2 e 3). Pressão intraocular de 12mmHg em ambos os olhos. À inspeção geral, presença de inúmeras manchas hipercrômicas do tipo “café-com-leite” distribuídas em tronco (Figura 4) e dorso (Figura 5).

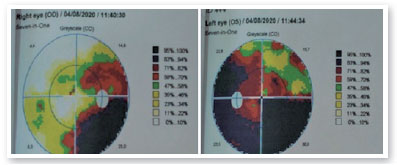
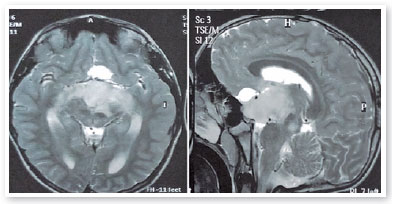
Solicitado propedêutica complementar com campo visual 24-2 que evidenciou, com boa confiabilidade, quadrantopsia inferior direita em AO (Figura 6), mais profunda em OE. À ressonância magnética de encéfalo, presença de volumosa formação tecidual expansiva de aspecto invasivo localizada na base do crânio com limites indefinidos e contornos irregulares medindo aproximadamente 3,9 a 4,7cm em seus maiores eixos ortogonais no plano sagital, envolvendo a haste, o infundíbulo hipofisário, os nervos ópticos e o quiasma óptico com extensão para a região núcleo-capsular esquerda e para o pedúnculo cerebral desse mesmo lado, altamente sugestiva de glioma de nervo óptico (Figura 7). Referenciado ao serviço de neurologia e oncologia pediátrica da Santa Casa de Belo Horizonte, onde optou-se por biópsia da lesão tumoral e subsequente confirmação diagnóstica de glioma de baixo grau ao anatomopatológico. Considerando-se a topografia da lesão e o elevado potencial de sequelas visuais em caso de exérese cirúrgica total, optou-se por seguimento quimioterápico com vincristina e carboplastina.
DISCUSSÃO
A neurofibromatose foi primeiramente publicada em 1768 como um relato de caso de um paciente portador de fibromas cutâneos supostamente herdados de seu pai4. Posteriormente, em 1882, Von Recklinghausen descreveu mais detalhadamente a patologia ao explicitar de forma mais complexa a origem nervosa dos tumores. Em 1940, Davis descreveu o glioma do nervo óptico associado à neurofibromatose5.
Trata-se de um designação genérica para um espectro de três doenças de origem genética autossômica dominante (AD), quais sejam: neurofibromatose tipo 1 (NF1); neurofibromatose tipo 2 (NF2) e schwannomatose. A NF1 - forma mais comum dentre as três - é causada pela alteração de um único gene localizado no cromossomo 176 gerando alteração na síntese da neurofibromina, uma proteína envolvida na diferenciação, controle da sobrevida, proliferação e mitogênese celulares do tecido nervoso e tegumento, tecido esquelético e sistema cardiovascular7. Essa proteína foi detectada por Nordlund et al. em todas as partes do encéfalo, sobretudo em neurônios com projeções mais extensas8.
Acomete aproximadamente 1/3000 nascidos vivos, sem preferência por sexo e é herdada por um dos pais em aproximadamente 50% dos casos. Nos demais casos não está relacionada à histórico familiar, o que sugere alta incidência de novas mutações1. As principais características clínicas da NF1 são as manchas café-com-leite (MCL), neurofibromas dérmicos e plexiformes, efélides axilares ou inguinais e os nódulos de Lish8.
Grande parte das crianças sintomáticas apresentam alterações oftalmológicas ao diagnóstico, desde baixa de acuidade visual a estrabismo; defeito pupilar aferente; atrofia de nervo óptico; papiledema e alteração na visualização de cores. Esses sintomas são frequentes nas faixas etárias inferiores a 7 ou 8 anos, sendo imprescindível que crianças com NF1 nessa faixa de idade sejam submetidas periodicamente a um exame oftalmólogico9.
Gliomas de nervo óptico estão entre os tumores mais frequentemente encontrados na NF110. Dentre eles, o astrocitoma é o mais frequente11. A associação de glioma das vias ópticas e NF1 geralmente é encontrada em crianças jovens, com menos de 10 anos de idade12. No caso específico desse relato, observou-se um extensão do tumor para além da região núcleo-capsular esquerdo e pedúnculo cerebral desse mesmo lado, justificando a baixa de visão percebida ao exame.
A NF1 possui diagnóstico essencialmente clínico através dos critérios diagnósticos do National Institutes of Healthy (NIH) para NF1, sendo altamente sensíveis e específicos para a grande maioria dos portadores da doença13 (Quadro 1).

Até o momento, não há nenhuma medicação específica disponível para reverter ou prevenir as lesões características da neurofibromatose. O tratamento deve ser avaliado caso a caso, levando-se em consideração a localização dos tumores, a sintomatologia e sua velocidade de crescimento. As opções terapêuticas incluem desde observação, quimio ou radioterapia até ressecção cirúrgica total ou parcial.
A NF1 é uma doença que pode se apresentar com vasta clínica oftalmológica, sendo que o oftalmologista é muitas das vezes o primeiro profissional a suspeitar de seu diagnóstico. Apesar de não apresentar tratamento ou prevenção específicos, seu diagnóstico precoce se faz importante para uma abordagem adequada e consequente preservação visual dos pacientes acometidos bem como para orientação e aconselhamento genético de seus familiares.
REFERÊNCIAS
1. Friedman JM. Epidemiology of neurofibromatosis type 1. Am J Med Genet. 1999;89(1):1-6.
2. Datson MM, Scrable H, Nordlund M, Sturbaum AK, Nissen LM, Ratner N. The protein product of neurofibromatosis type 1 gene is expressed at highest abundance in neurons, Schwann cells, and oligodendrocytes. Neuron. 1992;8(3):415-28.
3. Ferner RE, Huson SM, Thomas N, Moss C, Willshaw H, Evans DG, et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet. 2007;44(2):81-8.
4. Arkenside M. Observation on cancer. Trans Med Soc Lond. 1768; 1:64-92.
5. Geller M, Bonalumi AF. Neurofibromatose. In: Carakushansky G. Doenças genéticas em pediatria. 1° ed. Rio de Janeiro: Guanabara Koogan; 2001.
6. Barker D, Wright E, Nguyen K, Cannon L, Fain P, Goldgar D, et al. Gene for von Recklinghausen neurofibromatosis is in the pericentromeric region of chromosome 17. Science. 1987; 236(4805):1100-2.
7. Suchmacher M. Neurofibromatose tipo 1: revisão sistemática da literatura - 1a parte. Atualidades Médicas. 2018;2(4):171-81.
8. Norduland M, Gu X, Shipley MT, Ratner N. Neurofibrin is enriched in the endoplasmic reticulum of CNS neurons. J Neurosci. 1993;13(4):1588-600.
9. Tonsgard JH. Clinical manifestations and management of neurofibromatosis type 1. Semin Pediatr Neurol, 2006;13(1):2-7.
10. Stern J, DiGiacinto GV, Housepian EM. Neurofibromatosis and optic glioma: clinical and morphological correlations. Neurosurgery. 1979;4(6):524-8
11. Mukai K, Kitamura K, Asano N, Ohshima T, Hondo H, Matsumoto K. [Multifocal gliomas in cerebral hemisphere associated with von Recklinghausen’s disease: case report]. No Shinkei Geka. 1989;17(2):197-202.Japanese.
12. Bajenaru ML, Garbow JR, Perry A, Hernandez MR, Gutmann DH. Natural history of neurofibromatosis 1 associated optic nerve glioma in mice. Ann Neurol. 2005; 57(1):119-27.
13. Ferner RE, Gutmann DH. Neurofibromatosis type 1 (NF1): diagnosis and management. Handb Clin Neurol 2013;115:939-55.
INFORMAÇÃO DOS AUTORES
Financiamento: Declaram não haver
Conflitos de Interesse: Declaram não haver
Recebido em:
24 de Junho de 2021.
Aceito em:
25 de Agosto de 2021.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados



 Ler em inglês
Ler em inglês
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket