

Samilla Augusto Vieira de Araujo1; Renata Zaltron Neumann1; Vitor Gilberto Essi Monticuco2; Breno Reis Almeida2; Francyne Veiga Reis Cyrino3
DOI: 10.17545/eOftalmo/2021.0026
ABSTRACT
Leber congenital amaurosis (LCA) is an early-onset hereditary retinal dystrophy that becomes apparent in the first year of life and is characterized by severe visual impairment. Currently, 14 gene mutations that are associated with this phenotype have been identified and the transmission is autosomal recessive. We report a case of a 17-year-old male patient complaining of severe low visual acuity in both eyes since childhood, but a diagnosis had not been made. The patient did not have a family history of severe visual acuity. The family history was revealed that his parents were first cousins (consanguineous union). Fluorescein angiography, optical coherence tomography, electroretinography, and genetic testing were performed, which confirmed LCA diagnosis. Currently, studies on gene replacement therapy with promising results exist; however, they do not yet include all variants.
Keywords: Retinal Dystrophies; Amaurosis; Congenital Dystrophies; Retinal Degeneration.
RESUMO
A Amaurose Congênita de Leber (ACL) é uma distrofia retiniana hereditária de início precoce, do nascimento ao 1º ano de vida, caracterizada por deficiência visual severa. Atualmente estão identificados 14 genes cujas mutações se associam a este fenótipo e a transmissão é de modo autossômico recessiva. Relatamos um paciente do sexo masculino de 17 anos com queixa de baixa acuidade visual (BAV) severa desde a infância em ambos os olhos, sem diagnóstico e sem antecedentes familiares de baixa acuidade visual severa. Da história familiar destaca-se o fato dos pais serem primos consangüíneos de primeiro grau. Foi realizado angiofluoresceinografia (AGF), tomografia de coerência óptica (OCT), eletrorretinograma (ERG) e teste genético, confirmando o diagnóstico de amaurose congênita de Leber. Atualmente existem estudos com reposição de terapia genética com resultados promissores, mas que ainda não contemplam todas as variantes.
Palavras-chave: Distrofias da Retina; Amaurose; Distrofias Congênitas; Degeneração da Retina.
INTRODUCTION
Leber congenital amaurosis (LCA) is the term for a group of hereditary early-onset retinal dystrophies characterized by moderate to severe visual impairment that becomes apparent in the first months of life. Visual acuity, if assessed, ranges from 20/200 to no light perception and is rarely better than 20/2001.
In most cases, transmission is autosomal recessive, although, some cases are described with dominant transmission. Currently, 14 gene mutations associated with this phenotype have been identified.
During the first months of life, these patients develop nystagmus, high hyperopia, enophthalmos, their photomotor reflexes are often diminished or absent, and Franceschetti’s oculo-digital sign is common. Cataracts and keratoconus² may appear later in life. The following criteria are used in published studies to characterize patients with LCA: (1) Blindness or severe LVA in the first six months of life or first year of life3,4; (2) Absent or very reduced electroretinography (ERG )activity (photopic and scotopic)2,4-6; (3) Fundus of the eye appears normal or with minimal pigmentary changes5 and/or moderate arteriolar narrowing2,5; and (4) Absence of other multisystemic or retinal disorders4,6.
The findings in the fundus of the eye during the first months of life are normal or with minimal changes, which makes diagnosis difficult. ERG examination is the key to establishing the diagnosis²; little or no activity is detected in the retina, which indicates low visual function. It is common for patients to develop other changes in the retina over time, such as vascular attenuation, optic nerve atrophy (especially after the first year), pigmentary changes such as bone spicules, granular or colobomatous macular involvement, and pigmented nummular lesions at the posterior pole and in the periphery5.
In some cases, LCA is related to central nervous system complications, such as developmental delay, epilepsy, and impaired motor skills. Because LCA is relatively rare, the frequency of associated central nervous system complications is still unknown5. The differential diagnosis includes congenital stationary night blindness, achromatopsy, infantile retinitis pigmentosa, Joubert syndrome, Zellweger syndrome, and infantile Refsum edisease3.
CASE REPORT
A 17-year-old male student, E.F.D., complained of low visual acuity (LVA) in both eyes (OU) since early childhood. He reported involuntary eye movement (nystagmus) that began at six months of age when he was assessed and the hypothesis of Leber congenital amaurosis diagnosis was raised, but no additional tests were performed besides fundoscopy. He reported photophobia, visual difficulties at school, and some degree of learning deficit (using enlarged prints and sometimes rubbing the eye with fingers to see clearly). The patient had no family history of blindness or LVA. On examination, the patient exhibited bilateral horizontal nystagmus, corrected visual acuity of 20/200 in both eyes (OD: +2.50; S = −0.75 C 900° and OS: +3.50; S = −1.00 C 750°), biomicroscopy without abnormalities, intraocular pressure (IOP) of 10 mmHg in OU. Fundoscopy of OU showed macular coloboma and areas of pigmented epithelium hyperplasia in the coloboma area and in the periphery of the four quadrants (Figure 1).
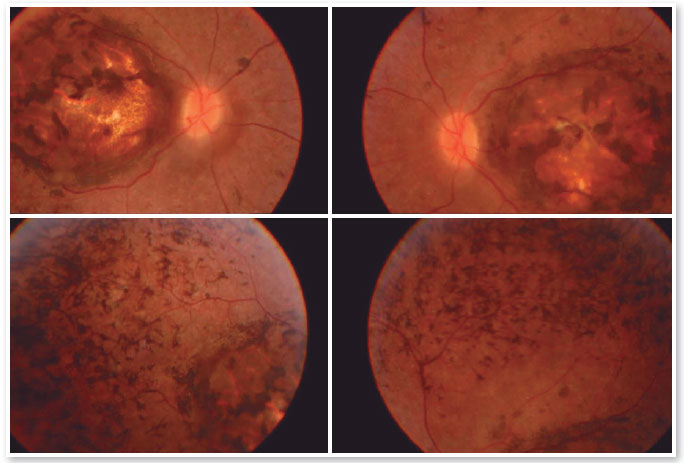
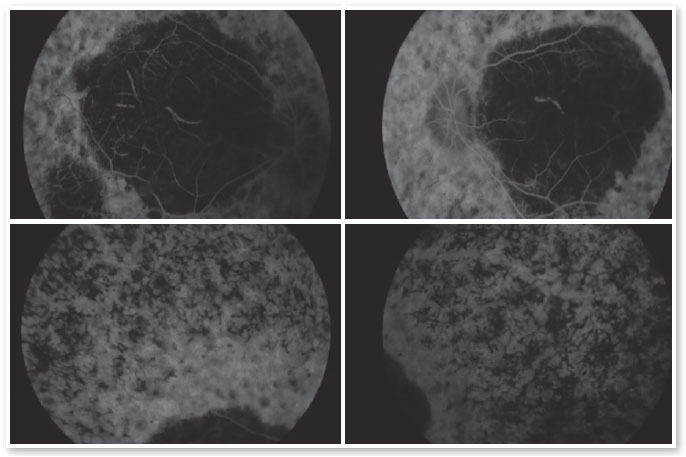
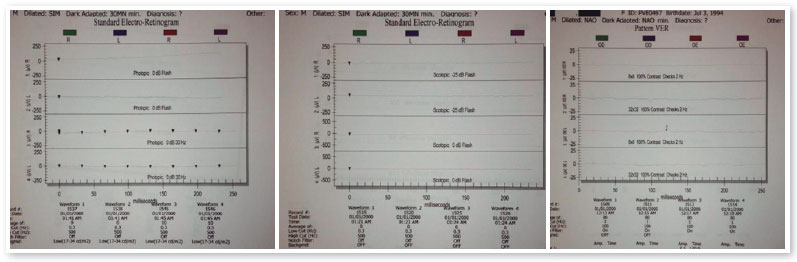
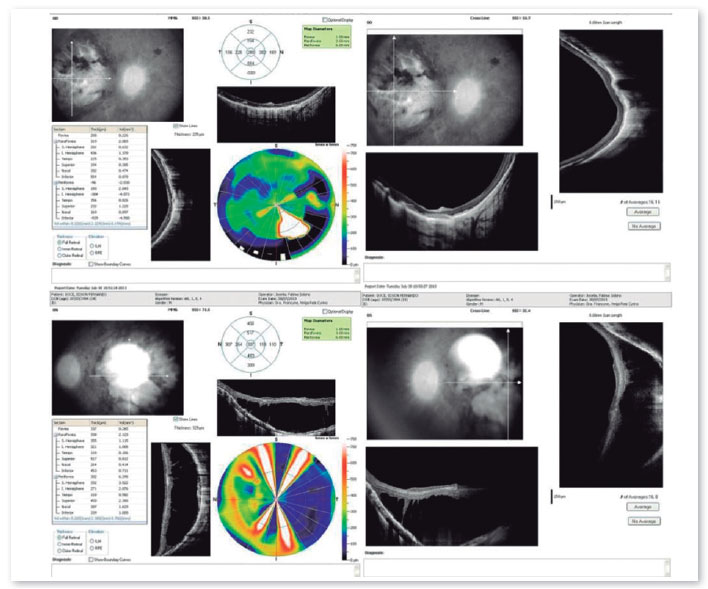
Complementary diagnostic tests were requested, which are listed in the figures below.
DISCUSSION
LCA is the most severe childhood hereditary dystrophy and its main feature is severe LVA at birth or in the first year of life. The diagnosis is made at approximately two years of age, when the symptoms of patients become apparent to parents, as in the case described above.
All patients with LCA have severe low vision or near blindness. Severe LVA is reported in all studies reported in the literature. The most common refractive error is hyperopia7, including in this case. The main symptoms are oculo-digital pressing, enophthalmos, strabismus, nystagmus, and photophobia1,3,4,8.
Fundoscopy findings have been described as normal or minimally altered in the early stages, with subsequent occurrence of changes of extremely variable appearance. The main test for the early diagnosis of LCA is ERG activity, which is absent or almost absent, even without changes in the fundus of the eye. A delay in neuropsychomotor development associated with LCA has been questioned and may result solely from sensory deprivation caused by the disease.
A proposal for gene replacement therapy for LCA has been studied for years. The medication voretigene neparvovec-rzyl (Luxturna) has been recently approved by the Food and Drug Administration and is indicated for children and adults with confirmed retinal dystrophy caused by biallelic mutations of the RPE65 gene, i.e., mutations in both copies of a gene inherited from the father and mother. The therapy works by introducing a normal copy of the gene into retinal cells using an adenovirus that transfers it to the nucleus of defective cells. This allows cells to start producing the protein that converts light into an electrical signal that can be transmitted by the optic nerve, effectively restoring vision9,10.
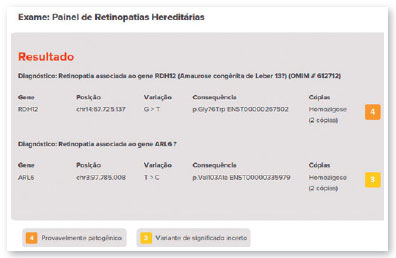
The therapy is applied only once in each eye with at least six days between surgical procedures. The most common adverse reactions of this treatment include conjunctival hyperemia, cataracts, and increased IOP. Approximately 1,000 to 2,000 people may have the mutation in the United States9,10. The genetic test did not show the mutation in the RPE65 gene in the case presented in this study. The detected variant was associated with the RDH12 gene, which is involved in the reduction of all-trans-retinal and its isomers (PMID: 15258582) and which, in this case, was inherited due to consanguinity of the parents (family variant).
In cases of hereditary dystrophies, genetic counseling is mandatory to reduce the possibility of transmission in the following generations. In the above-described case, the mutation will not be transmitted if the patient does not marry within the family. Unfortunately, to the best of our knowledge, no treatment has been reported for this variant; however, studies are underway and it will be possible to slow and even halt the progression of these debilitating conditions in the not too distant future.
ACKNOWLEDGEMENTS
We thank Dr. Juliana Salum who was the intermediator for conducting the genetic test provided free of charge by Mendelics, and Dr. Mario Ogata for the patient’s referral and follow-up.
REFERENCES
1. Steinberg A, Ronen S, Zlotogorski Z, Silverston BZ, Hirsch I, Nawratzki. J PediatrOphthalmolStrabismus1992;29:2
2. Schroeder R, Mets MB,Maumenee IH; Leber’s congenital amaurosis. Retrospective review of 43 cases and a new fundus finding in two cases. ArchOphthalmol 1987;105(3):356-9.
3. Leber congenital amaurosis – researchadvances - http://www.blindness.org/leber-congenital-amaurosis acessado em 19/04/2018
4. Vaizey MJ, Sanders MD, Wybar KC, Wilson J. N. Neurological abnormalities in congenital amaurosis of Leber – 1977
5. Nickel B, Hoyt CS. Leber’s congenital amaurosis - is mental retardation a frequent associated defect? - 1982
6. Steinberg A, Ronen S, Zlotogorski Z, Silverston BZ, Hirsch I, Nawratzki. J PediatrOphthalmol Strabismus 1992;29:224-227
7. Lambert SR, Taylor D, Kriss A. The infant with nystagmus, normal appearing fundi, but an abnormal ERG. SurvOphthalmol 1989a;34(3):173-186.
8. Casteels I, Spileers W, Demaerel P, Casaer P. De Cook P., Dralands L, Missotten L. Neuropediatrics 1996; 27:183-193
9. Noble KG, Carr RE. Leber congenital amaurosis. A retrospective study of 33 cases and an histopatological study of one case. ArchOphthalmol 1978; 96:818-821
10. Schroeder R, Mets MB,Maumenee IH; Leber’s congenital amaurosis. Retrospective review of 43 cases and a new fundus finding in two cases. ArchOphthalmol 1987;105(3):356-9.
11. Dekaban A. Hereditary syndrome of congenital night blindness (Leber), polycystic kidneys and maldevelopment of the brain. AM. J. Ophthalmol1969;68:1029-1037. Apud c Nnickel e Hoyt.
12. Fulton AB, Hansen RM, Mayer L; Vision in Leber Congenital Amaurosis. ArchOphthalmol 1996; 114 (6): 698-703.
13. Lambert SR, Kriss A, Taylor D, Coffrey R, Pembey M. Follow up and diagnostic reappraisal of 75 patients with Leber’s congenital amaurosis. 1989b
14. Fishman GA. Electrophysiologic testing in disorders of the retina, optic nerve and visual pathway. Ophthalmology Monographs. The Foundation of the American Academy of Ophthlamology. 2001.308p.
16. Luxturna [prescribing information]. Philadelphia, PA; Spark Therapuetics, Inc. December 2017.
AUTHOR’S INFORMATION




*Residentes do Centro Avançado em Oftalmologia- Universidade de Ribeirão Preto – CAO-UNAERP.
** Médico oftalmologista.
** Preceptora da residência de oftalmologia do CAO-UNAERP, medica assistente do HCFMRP-USP Ribeirão.
Funding: No specific financial support was available for this study
Disclosure of potential conflicts of interest: None of the authors have any potential conflict of interest to disclose
Received on:
July 21, 2020.
Accepted on:
December 15, 2020.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados

 Ler em português
Ler em português
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket