

Samilla Augusto Vieira de Araujo1; Renata Zaltron Neumann1; Francyne Veiga Reis Cyrino1,2,3
DOI: 10.17545/eOftalmo/2021.0006
ABSTRACT
Retinitispigmentosa (RP) is a hereditary dystrophy of the retina in which progressive destruction of the neurosensory retina occurs. Although unilateral RP was first described more than a century ago, it is not yet a well-defined entity. This is attributed to the difficulty of ruling out secondary causes,primarily infectious ones. In the presentcase, the patient presented a clinical picture similar to unilateral RPbut with laboratory tests and clinical history that corroborated the hypothesis of pseudo-retinitis pigmentosa secondary to syphilis. Although there is no specific treatment for thiscase, its discussion becomes important because of the current exponential growth of syphilis.
Keywords: Retinitis pigmentosa; Syphilis; Retinal degeneration; Retinal photoreceptor cells.
RESUMO
A retinose pigmentar é uma distrofia hereditária da retina em que ocorre a destruição progressiva da retina neurossensorial. Os casos unilaterais, retinose pigmentar unilateral, apesar de ter sido descrito pela primeira vez há mais de um século ainda não é uma entidade bem definida. Isso ocorre pela dificuldade de descartar causas secundárias principalmente as infecciosas. No caso em questão, o paciente apresentou quadro semelhante a retinose pigmentar unilateral porém com exames laboratoriais e história clínica que corroboraram com a hipótese de Pseudoretinose Pigmentar secundária a Sífilis. Apesar de não se ter um tratamento específico para o caso a discussão se torna importante devido ao crescimento exponencial da Sífilis atualmente.
Palavras-chave: Retinose pigmentar; Sífilis; Degeneração retiniana; Células fotorreceptoras retinianas bastonetes.
INTRODUCTION
Retinitis pigmentosa (RP) is an umbrella term used to characterize certain diseases in which progressive destruction of retinal photoreceptors occurs, particularly rods, responsible for light-dark adaptation.This is the earliest symptom of the disease and is called nyctalopia. RPis the most common hereditary dystrophy (1:5000) and is usually diagnosedwhile patients are still at a young age. Most cases are bilateral2,3 and associated with a mutation in the RPE65 gene.
As rod degeneration progresses, the clinical image worsens, with consequent loss of peripheral vision and visual field constriction, progressively evolving to macular impairment, which may cause visual impairment, macular edema, posterior subcapsular cataracts, glaucoma, and even blindness2. Anatomically,its classic signs are a triad of retinal pigmentation shaped such as bone spicules, “waxy” pallor, and arteriolar attenuation2,3.
Diagnosis is based on the clinical history and the changes reported in fundus biomicroscopy. Complementary examinations, such as visual field measurement, show peripheral scotomas evolving to a tubular pattern. Electroretinography shows a reduction in photoreceptor function,particularly in rods. Fluorescein angiography is important for monitoring the condition, revealing the degree of destruction of the pigment epithelium.
Although unilateral retinitis pigmentosa (URP) was described more than a century ago, it is not yet a well-defined entity3. Certain cases have been reported, but it is difficult to exclude secondary diseases that present the same unilateral picture4. In 1952, François and Verriest established criteria for the correct diagnosis, according to which anassociation with infectious diseases or trauma should be excluded, and the presence of retinitis in the affected eye and the absence of signs in the contralateral eye must be attested5.
The aim of the presentstudy is to report a case of infectious unilateral pseudo-retinitis pigmentosa (PRP)without an adequate diagnosis.
CASE REPORT
A male patient, 72-year-old, smoker, rural worker, attended a consultation at the Outpost for Advanced Ophthalmology of the University of Ribeirão Preto (CAO-UNAERP), Brazil,complaining of progressive loss of near vision in both eyes (OU) for ~2 years, more pronounced in the left eye (OS). He reported an electric welding accident injuring the OS>20 years before, with no visual deficit at the time. He denied known comorbidities or family history of eye diseases and blindness.
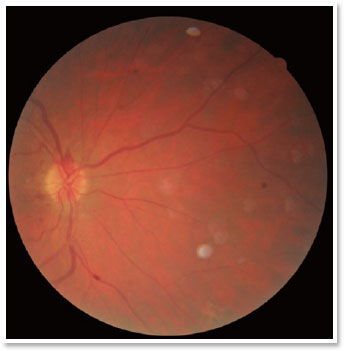
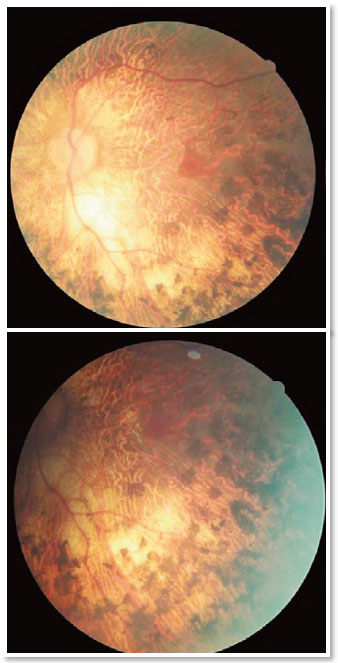
A complete ophthalmological examination was performed with corrected visual acuity of 20/30 in the right eye (OD) and 20/25 in the OS, a nuclear cataract 2+/4+, and tonometry of 13mmHg in OU. At fundoscopy, the OD showed no changes (Figure 1); however, in the OSan optic disc with sharp margins could be observed,as well as a markedly pale papilla, vascular attenuation, and the presence of pigmentary lesions with the appearance of bony spicules diffusedthroughout the posterior pole and periphery, but sparing the macular region (Figure 2).These results suggested a probable diagnosis of URP.
Given these fundoscopicresults, the patient was questioned again about his clinical history and reported risky sexual behavior as a young adult and even after marriage.His wife reported that the patient had been treated with intramuscular penicillin a few years earlier.
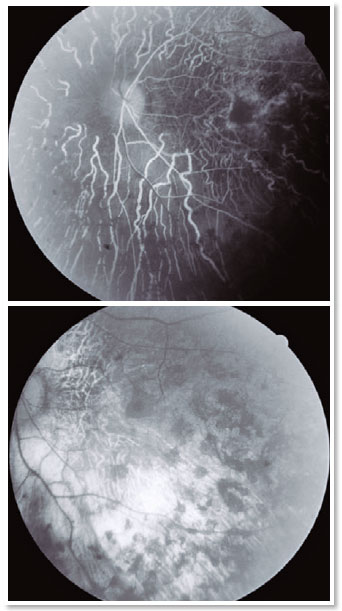
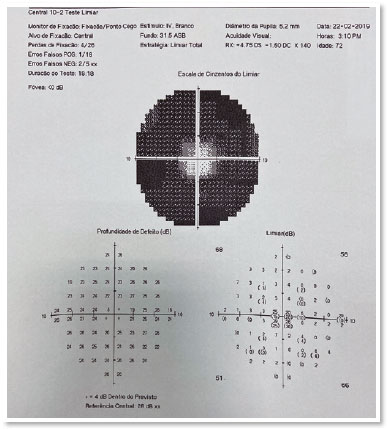
Fluorescein angiography (FA), optical coherence tomography (OCT) (Figure3), and a visual field test (Figure 4) were requested. The visual field test was normal in the OD,but a tubular field with a central vision island was evidenced in the OS.FA showed diffuse hypofluorescentareas of blockade mottled by window-effect hyperfluorescence in the four quadrants and in the macular region (Figure 3). OCT did not show any macular edema in the OS (Figure 4).
VDRL was non-reagent (1:1); other serology tests (FTA–ABS, toxoplasmosis, and cytomegalovirus) were all IgM-negative and IgG-positive.
DISCUSSION
Infectious diseases, such as syphilis, toxoplasmosis, rubella, and cytomegalovirus infection, can cause an inflammatory reaction leading to a mobilization of the retinal pigment epithelium (RPE)1,6 and the formation of bone spicule-shaped pigmented areas that are characteristic of RP. Other possible causes, such as eye trauma associated with retinal detachment7 ortoxic retinitis secondary to the use of medications (such as antipsychotics) are significant and must be considered in the differential diagnosis8.
In the presentstudy, despite the recent (two years)complaint of worsening visual acuity and the marked presence of fundoscopic changes only in the OS,the patient’s history of risky sexual behavior and his good visual acuity suggested the hypothesis of PRP secondary to an infectious disease (syphilis). According to the literature, secondary conditions such as syphilitic PRP generally present more favorable visual prognosis than cases of hereditary RP, despite the risk of optic neuritis, which can lead to total loss of visionif not treated in time.
Until recently, there was no specific treatment for these disorders of the retinal epithelium. Certain studies reported onthe use of oral vitamin A for RP, but its effect was not proven beyond side effects9. The use of valproic acid was proposed in an attempt to slow the progression of the visual field, but no benefit was reported when compared to placebo8. Recently, voretigene neparvovec-rzyl (Luxturna®) was approved in the United States for treating RP. The candidates for this new treatment would be those patients who have aproven biallelic mutation in the RPE65 gene and who still have sufficient viable retinal cells in the OCT10. Applied via subretinal injection, this medication uses a modified virus to introduce a normal copy of the gene into the retinal cells, causing the defective cells to produce the protein that converts light into a sensory signal conducted by the optic nerve10,11.
For secondary PRP, the cause must be treated, which for syphilis would mean the use of benzathine or crystalline penicillin at the recommended doses and routes of administration. However, for the patient in question, no specific treatment was instituted, following the guidance of an infectiologist because the patient had already been previously treated with intramuscular penicillin. So, a biannual follow-up was selected.
There are numerous possible causesof cell injury that canlead to mobilization of the pigment epithelium, photoreceptor injury, and pigment deposition similar to the case described. The existence of isolated URPasa clinical entity is controversial, primarily because of the difficulty of ruling outsecondary causes, including syphilis, whose incidence in both young and elderly people has been increasing greatly in Brazil. However, a thoroughly collected clinical history can guide the reasoning and diagnosis.
REFERENCES
1. Tayah D, Angelucci RI, Sampaio P, Rehder JRCL. Retinose Pigmentar. Arq Med ABC. 2004;29:82-6.
2. Bowling B. Kanski Oftalmologia Clínica: Uma Abordagem Sistêmica. GEN Guanabara Koogan ed. 2016.
3. Rios DFC, Carneiro LFSA, Cunha AAF, Dalle CBM, Frasson M. Retinose pigmentar unilateral ou pseudorretinose pigmentar?: relato de caso. Arq Bras Oftalmol. 2013;76(6):383-5.
4. Vela JI, Marcantonio I, Díaz-Cascajosa J, Crespí J, Buil JA. Progression of retinal pigmentation mimicking unilateral retinitis pigmentosa after bilateral pars planitis: a case report. BMC Ophthalmol. 2018;18(1):242.
5. François J, Verriest G. Rétinopathiepigmentaire unilateral. Ophthalmologica. 1952;124(2):65-88
6. Aragão REM, Barreira IMA, Bastos ASV, Carneiro GJAM, Oria TPC. Unilateral retinitis pigmentosa: case report. Rev Med UFC. 2015;55(2):54-8.
7. Duncan JL, Pierce EA, Laster AM, Daiger SP, Birch DG, Ash JD, Iannaccone A, Flannery JG, Sahel JA, Zack DJ, Zarbin MA, and the Foundation Fighting Blindness Scientific Advisory Board. Inherited Retinal Degenerations: Current Landscape and Knowledge Gaps. Transl Vis Sci Technol. 2018;7(4):6.
8. Cestari AT, Sallum JMF, Conti ML, Tagliari TI, Barboza MNC. Retinose pigmentada unilateral secundária a trauma: relato de caso. Arq Bras Oftalmol. 2012;75(3):210-2 .
9. Weller JM, Michelson G, Juenemann AG. Unilateral retinitis pigmentosa: 30 years follow-up. BMJ Case Rep. 2014 Feb 10;2014: bcr2013202236.
10. Luxturna [prescribing information]. Philadelphia, PA; Spark Therapuetics, Inc. December 2017.
11. LuxturnaTM (Voretigene Neparvovec-Rzyl) UnitedHealthcare Commercial Medical Benefit Drug Policy Effective 11/01/2019 Proprietary Information of UnitedHealthcare. Copyright 2019
AUTHOR’S INFORMATION



Funding: No specific financial support was available for this study
Disclosure of potential conflicts of interest: None of the authors have any potential conflict of interest to disclose
Received on:
September 26, 2020.
Accepted on:
December 15, 2020.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados
 Ler em português
Ler em português
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket