

Roberta de Julio Matheus; Julia Alves Utyama; Guilherme Novoa Colombo Barboza; Marcello Novoa Colombo Barboza; Juliana Carolina Contrera
DOI: 10.17545/eOftalmo/2021.0005
ABSTRACT
Usher syndrome is an autosomal recessive disease with ophthalmological involvement, characterized by retinitis pigmentosa, and otological impairment, characterized by neurosensory deafness. It can be classified into three types, according to the time of onset and the severity of symptoms. Patients with this disease present progressive visual loss, the first symptom being nyctalopia, followed by loss of the peripheral visual field. Vestibular disorders, such as ataxia, may be associated. The diagnosis of the syndrome is important for conducting genetic counseling and for initiating early visual rehabilitation to improve the patient’s quality of life.
Keywords: Usher syndrome; Retinitis pigmentosa; Nyctalopia.
RESUMO
A Síndrome de Usher é uma doença autossômica recessiva com acometimento oftalmológico, caracterizado por Retinose Pigmentar, e otorrinolaringológico, caracterizado por surdez neurossensorial. Pode ser classificada em 3 tipos variando de acordo com o início e gravidade dos sintomas. Pacientes com essa doença apresentam perda visual progressiva, sendo o primeiro sintoma a cegueira noturna seguida de perda de campo visual periférico. Distúrbios vestibulares, como ataxia, podem estar associados. O diagnóstico da doença é importante para realização de aconselhamento genético e para iniciar reabilitação visual precoce para a melhoria da qualidade de vida do paciente.
Palavras-chave: Síndromes de Usher; Retinose pigmentar; Cegueira noturna.
INTRODUCTION
Usher syndrome is an autosomal recessive disease1 characterized by neurosensory hypoacusis of varying degrees and retinitis pigmentosa (RP) 2. It has an incidence of 3 in every 100,000 individuals in the general population2, 3%-6% in the population of hearing-impaired persons2,3, and ~50% of cases of hereditary deafness1. It can be classified into three distinct types that differ based on the time of onset and the severity of symptoms1,2. Type 1 is characterized by total congenital deafness, vestibular impairment, and nyctalopia in childhood3,4. Type 2 shows partial congenital deafness, vestibular dysfunction, and nyctalopia in young adults3. Type 3 is characterized by a progressive hearing deficit without balance problems and with visual changes in adult life3.
RP is present in all types of the disease, differing in the age of onset2,5. It appears earliest in Type 1, namely, in the first decade of life5. The most significant initially noticed symptom, present in all types, is nyctalopia, which slowly expands to alterations in peripheral vision4. In these patients’ fundoscopy images, it is possible to observe typical RP findings such as pallor of the optic nerve, arteriolar sclerosis, and migration of intraretinal pigments in the form of bone spicules5. Subcapsular cataracts, optic nerve drusen, atrophic foveal lesions, and cystoid macular edema (CME) may be present, the latter being most commonly evidenced with optical coherence tomography (OCT)5.
Complementary examinations, such as visual field (VF) tests and electroretinography (ERG), may be useful in cases with strong diagnostic suspicion but absent ophthalmologic signs5. In VF tests, the loss of sensitivity can be observed in different phases of the disease; however, this result is nonspecific. ERG is a complex record of retinal electrical potentials in response to light stimulation, and these potentials are generally reduced or absent in patients with this syndrome6. ERG may show retinal changes that precede fundoscopic changes, and it is the examination of choice for diagnosis5.
This study aims to report a case of Usher Syndrome and to use ophthalmic findings as an aid to classify the disease.
CASE REPORT
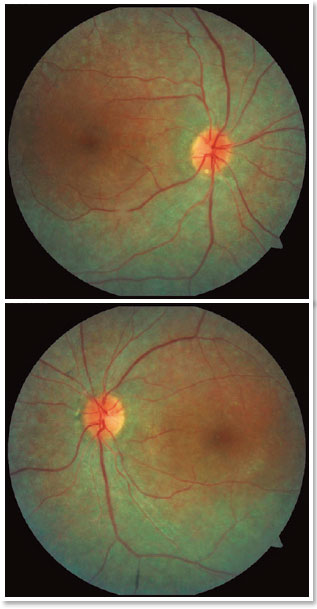
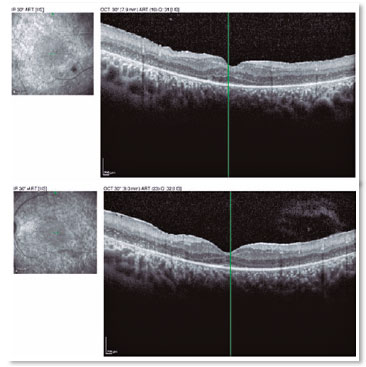
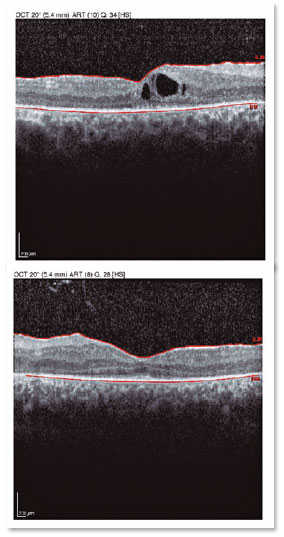
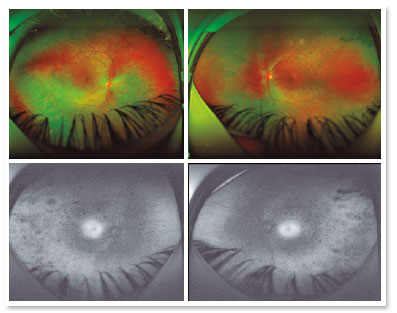
A male patient, initials R.T.M., 23 years old, white, student, complained of low far and near visual acuity (VA) for 20 years, with progressive worsening for five years and peripheral vision impairment, particularly intense at night. Personal background: delayed psychoneuromotor development, gait alteration (ataxia), Usher syndrome diagnosed in 2007 through ERG, preverbal deafness, and a cochlear implant received in 2014. Ophthalmic history: ERG report (2007) showing undetectable rod responses in both eyes (OU) rods and 95% reduced cone responses OU. Family history: paternal sister with unilateral blindness without apparent cause. On examination: corrected VA: 20/200 OU. Biomicroscopy without changes. Intraocular pressure: right eye (OD) 13mmHg and left eye (OS) 14mmHg. Fundoscopy: physiological excavation OU. Fundoscopy: normally colored optic nerve, physiological excavation, preserved vessels, mobilization of the retinal pigment epithelium (RPE), multiple diffusely distributed hypopigmented spots preserving the macula OU (Figure 1). OCT demonstrated an irregularity in the ellipsoid zone and preserved features in the foveal region, presence of a hyper-reflective structure above the internal limiting membrane (ILM), characterizing an epiretinal membrane (ERM), a thinning of the parafoveal external nuclear layer OU (Figure 2), and the presence of intraretinal hyporeflective cystic cavities evidencing macular edema in OD (Figure 3). Optomap evidenced hyperpigmented areas (“bone spicules”) in the periphery OU (Figure 4). The patient underwent evaluation by the clinic’s Retina Department, which recommended an intravitreal injection of dexamethasone (Ozurdex®); however, because of the patient’s mobility restrictions because of the syndrome, he did not return to the service, which made it impossible to evaluate his clinical evolution using medication.
DISCUSSION
Usher syndrome is characterized by neurosensory hypoacusis associated with progressive visual loss. Its primary initial visual symptoms are nyctalopia and loss of peripheral visual field. When analyzing this case, it can be concluded with certainty that the patient presents type 1 Usher syndrome because of the progressive visual changes in early adult life, associated with preverbal deafness and gait alteration, as described below.
Correct diagnosis is important because measures to improve quality of life and genetic counseling should be performed as soon as possible2,6. Diagnosing the syndrome only via clinical results can be challenging, requiring a molecular genetics analysis2 and complementary examinations such as ERG, OCT, and autofluorescence retinography. To date, eleven genes associated with the disease have already been identified7.
ERG evaluates the response of photoreceptors to light stimuli through electrodes in contact with the cornea; moreover, it is of great importance for assessing the loss of function of retinal structures because it can provide information on the prognosis and response to treatment8. The most relevant results in patients with Usher syndrome is the reduced or absent response of cones and rods6. This characteristic is attributed to the frequent and severe involvement of the external nuclear layer of the retina, which contains the photoreceptor nuclei, in patients with RP9. However, the internal nuclear layer of the retina, which is composed of amacrine, bipolar, and horizontal cells, is normally preserved in this disease; nevertheless, it may present damage, depending on the evolution of the syndrome9. In most cases, the loss of function of rods is greater than that of the cones9, as observed in the reported patient. Note that this ERG result often precedes fundoscopic alterations8 and clinical changes because there may be a 90% loss of cone function while the patient’s visual acuity remains good. Therefore, for early diagnosis, objective examinations such as ERG are better 9.
OCT is non-invasive and useful for evaluating retinal morphology, particularly of the macula, as OCT can measure the retinal thickness, evaluate the quality of the photoreceptor layer, and determine the presence of CME9 and ERM3. Both CME and ERM occur because of the inflammatory process present in Usher syndrome, which may alter RPE permeability, leading to these results. Hyporeflective intraretinal lesions compatible with the presence of a fluid and foveal thickening are observed in OCT, as well as a hyperreflective membrane above the ILM, characterizing CME and ERM respectively8. Because OCT evaluates the status of the photoreceptor layer, this examination is fundamental to predict the impact of treatment on the evolution of the disease and consequently on these patients’ prognosis9.
Retinography with autofluorescence is useful to document the deterioration of the RPE, which is characterized by local accumulation of lipofuscin, starting at the periphery of the retina8,9. In this examination, hyperautofluorescent areas produce the lowest amplitudes in the ERG; therefore, they are the sites with the greatest local RPE damage9 and can be frequently observed in these patients.
The ophthalmological management of Usher syndrome comprises attenuating and preventing the progression of RP, thereby improving the patient’s quality of life. Currently, there are multiple ongoing studies in the literature on new therapeutic strategies such as artificial retinal implants10,11, pharmacological therapy12,13, and gene therapy14.
Artificial retinal implants comprise either microchips that come into contact with the photoreceptor layer15-17 or electrodes implanted in the inner part of the retina in contact with ganglion cells. Significant improvements in the ability to read and recognize objects have already been reported in those cases18. There are ongoing studies evaluating the possibility of transplantation of RPE, photoreceptors19, or stem cells20, with promising results such as increased VA21 and protection of retinal neurons22.
Regarding the pharmacological approach, there is evidence of vitamin A supplementation resulting in a slower loss of the visual field9; however, periodic monitoring should be maintained to evaluate possible side effects such as osteoporosis. Docosahexaenoic acid, an omega-3 fatty acid, has been used for treatment because rhodopsins and iodopsins contain high levels of this substance and patients with RP have lower serum levels of it9.
For treating CME, the intravitreal injections of corticosteroids or anti-vascular endothelial growth factors (anti-VEGF) such as bevacizumab may be indicated8, in addition to oral or topical carbonic anhydrase inhibitors, which can provide a transient improvement in VA in these patients23-25. Measures that improve VA, such as cataract extraction and reduced exposure to light, can be beneficial9. In more advanced cases, optical aids for subnormal vision, such as magnification in electronic devices such as laptop computers and tablets, can improve the patient’s quality of life26,27.
In terms of gene therapy, a significant restoration of vision in nearly blind young patients has been evidenced. However, additional studies should be performed to improve these treatments’ long-term biocompatibility and stability18,26,28.
When evaluating the reported case, it was possible to observe Usher syndrome’s characteristic clinical pattern: a young adult patient, initial RP symptoms associated with neurosensory deafness, and gait changes. Multiple patients end up abandoning treatment because of mobility-related difficulties and the modest improvement observed with available treatments. However, despite the syndrome’s progressive nature and reserved visual prognosis, the technological advancement of the available treatments has been able to improve the patients’ quality of life. Therefore, it is extremely important to encourage them to maintain regular ophthalmological and otological monitoring.
REFERENCES
1. Bonnet C, El-Amraoui A. Usher syndrome (sensorineural deafness and retinitis pigmentosa): pathogenesis, molecular diagnosis and therapeutic approaches. Curr Opin Neurol. 2012;25(1):42-9.
2. Benson, MD, MacDonald M. Bilateral uveitis and Usher syndrome: a case report. J Med Case Rep. 2015 Mar 15;9:60.
3. Liarth JCS, Gonçalves EA, Gonçalves JOR, Neiva DM, Leal FAM. Síndrome de Usher: características clínicas. Arq Bras Oftalmol. 2002;65(4):457-61.
4. Testa F, Melillo P, Rossi S, Marcelli V, Benedicts A, Colucci R, et al. Prevalence of macular abnormalities assessed by optical coherence tomography in patients with Usher syndrome. Ophtalmic Genetics. 2017;39(1):17-21.
5. Friedman TB, Schultz JM, Ahmed ZM, Tsilou ET, Brewer CC. Usher Syndrome: Hearing Loss with Vision Loss. Adv Otorhinolaryngol. 2011;70:56-65.
6. Mendieta L, Berezovsky A, Salomão SR, Sacai PY, Pereira JM, Fantini SC. Acuidade visual e eletrorretinografia de campo total em pacientes com síndrome de Usher. Arq Bras Oftalmol. 2005; 68(2):171-6.
7. Jouret G, Poirsier C, Spodenkiewicz M, Jaquin C, Gouy E, Arndt C, et al. Genetics os Usher Syndrome: New Insights From a Meta-analysis. Otol Neurotol. 2019;40(1):121-9.
8. Buchaim G, Rezende MP, Maia M. Implante intravítreo de liberação crônica de dexametasona (Ozurdex®) para o tratamento de edema macular por retinose pigmentar: relato de caso. Arq Bras Oftalmol. 2013;76(6):377-9.
9. Hartong DT, Berson EL, Dryja TP. Retinitis Pigmentosa. Lancet. 2006 Nov 18;368(9549):1795-809.
10. Djilas M, Oles C, Lorach H, Bendali A, Dégardin J, Dubus E, et al. Three-dimensional electrode arrays for retinal prostheses: modeling, geometry optimization and experimental validation. J Neural Eng. 2011;8(4):046020.
11. Wilke R, Gabel VP, Sachs H, Schmidt KUB, Gekeler F, Besch D, et al. Spatial resolution and perception of patterns mediated by a subretinal 16-electrode array in patients blinded by hereditary retinal dystrophies. Invest Ophthalmol Vis Sci. 2011;52(8):5995- 6003.
12. Genead MA, Fishman GA. Efficacy of sustained topical dorzolamide therapy for cystic macular lesions in patients with retinitis pigmentosa and Usher syndrome. Arch Ophthalmol. 2010;128(9):1146-50.
13. Tao W. Application of encapsulated cell technology for retinal degenerative diseases. Expert Opin Biol Ther. 2006;6(7):717-26.
14. Zou J, Luo L, Shen Z, Chiodo VA, Ambati BK, Hauswirth W, et al. Whirlin replacement restores the formation of the USH2 protein complex in whirlin knockout photoreceptors. Invest Ophthalmol Vis Sci. 2011;52(5):2343-51.
15. Zrenner E. Will retinal implants restore vision? Science. 2002; 295(5557):1022-5.
16. DeMarco PJ Jr, Yarbrough GL, Yee CW, McLean GY, Sagdullaev BT, Ball SL, et al. Stimulation via a subretinally placed prosthetic elicits central activity and induces a trophic effect on visual responses. Invest Ophthalmol Vis Sci. 2007;48(2):916-26.
17. Zrenner E, Bartz-Schmidt KU, Benav H, Besch D, Bruckmann A, Gabel VP, et al. Subretinal electronic chips allow blind patients to read letters and combine them to words. Proc Biol Sci. 2011; 278(1711):1489-97.
18. Berger AS, Tezel TH, Del Priore LV, Kaplan HJ. Photoreceptor transplantation in retinitis pigmentosa: short-term follow-up. Ophthalmology. 2003;110(2):383-91.
19. Seiler MJ, Aramant RB. Transplantation of neuroblastic progenitor cells as a sheet preserves and restores retinal function. Semin Ophthalmol. 2005;20(1):31-42.
20. Radtke ND, Aramant RB, Seiler MJ, Petry HM, Pidwell D. Vision change after sheet transplant of fetal retina with retinal pigment epithelium to a patient with retinitis pigmentosa. Arch Ophthalmol. 2004;122(8):1159-65.
21. Meyer JS, Katz ML, Maruniak JA, Kirk MD. Embryonic stem cell-derived neural progenitors incorporate into degenerating retina and enhance survival of host photoreceptors. Stem Cells. 2006;24(2):274-83.
22. Fishman GA, Gilbert LD, Fiscella RG, Kimura AE, Jampol LM. Acetazolamide for treatment of chronic macular edema in retinitis pigmentosa. Arch Ophthalmol. 1989;107(10):1445-52.
23. Chen JC, Fitzke FW, Bird AC. Long-term eff ect of acetazolamide in a patient with retinitis pigmentosa. Invest Ophthalmol Vis Sci. 1990;31(9):1914-8.
24. Reis RF, Moreiro-Gonçalves N, Silva SE, Brandão EM, Falcão-Reis FM. Comparison of Topical Dorzolamide and Ketorolac Treatment for Cystoide Macular Edema in Retinitis Pigmentosa and Usher’s Syndrome. Ophthalmologica. 2015;233(1):43-50.
25. Berson EL, Rabin AR, Mehaff ey L III. Advances in night vision technology: a pocketscope for patients with retinitis pigmentosa. Arch Ophthalmol. 1973;90(6):427-31.
26. Stieglitz T. Development of a micromachined epiretinal vision prosthesis. J Neural Eng. 2009;6(6):065005.
27. Busskamp V, Duebel J, Balya D, Fradot M, Viney TJ, Siegert S, et al. Genetic reactivation of cone photoreceptors restores visual responses in retinitis pigmentosa. Science. 2010;329(5990):413-7.
AUTHOR’S INFORMATION




Funding: No specific financial support was available for this study
Disclosure of potential conflicts of interest: None of the authors have any potential conflict of interest to disclose
Received on:
January 24, 2020.
Accepted on:
August 17, 2020.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados

 Ler em português
Ler em português
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket