

Natália Maia de Faria1; Lívia Freire Reis2; Josué Geraldo Lessa3
DOI: 10.17545/eoftalmo/2018.0036
ABSTRACT
INTRODUCTION: Bardet-Biedl syndrome is a rare autosomal recessive disease with the chief clinical manifestations of polydactylism; central obesity; mental retardation; hypogenitalism; and retinal dystrophy, which leads to progressive visual loss since childhood.
CASE REPORT: A 17-year-old boy presented with progressive low visual acuity in both eyes. He had cardiac malformation since birth, intellectual disability, divergent strabismus, short stature, obesity, and polydactyly of hands and feet. Ophthalmologic examination using Snellen chart revealed a visual acuity of 0.1 in both eyes and normal intraocular pressure and biomicroscopy result. Fundoscopy revealed discoloration of the retinal pigment epithelium, giving the fundus a grayish color and mottled appearance; a decrease in vessel diameter; paleness of the optic papilla; deposits of macular and peripheral bone spicule-shaped pigments; and increase in foveal reflex.
CONCLUSION: Retinal dystrophy, which occurs in approximately 90% of patients with Bardet-Biedl syndrome, usually confirms its diagnosis. This manifestation indicates the importance of the role of an ophthalmologist in the initial investigation and follow-up of systemic complications.
Keywords: Retinal Dystrophies; Retinitis Pigmentosa; Bardet-Biedl Syndrome.
RESUMO
INTRODUÇÃO: A síndrome de Bardet-Biedl é uma doença de herança autossômica recessiva rara cujas principais manifestações clínicas são polidactilia, obesidade, retardo mental, hipogenitalismo e distrofia retiniana que causa progressiva perda visual a partir da infância.
RELATO DE CASO: Paciente, 17 anos, sexo masculino, apresentou baixa acuidade visual progressiva em ambos os olhos. Apresentava déficit intelectual e má formação cardíaca desde o nascimento, assim como estrabismo divergente. Além disso, tinha baixa estatura, obesidade e polidactilia de mãos e pés. Ao exame oftalmológico, constatou-se acuidade visual com correção de 0,1 pela tabela de Snellen em ambos os olhos, pressão intraocular e biomicroscopia normais. À fundoscopia, observou-se descoloração do epitélio pigmentado da retina dando ao fundo uma coloração acinzentada e um aspecto mosqueado, diminuição do calibre vascular, presença de palidez de papila, migração de pigmento em padrão de espículas ósseas, inclusive macular, e aumento do reflexo foveal.
CONCLUSÃO: A distrofia retiniana está presente em cerca de 90% dos casos já descritos na síndrome de Bardet-Biedl e muitas vezes é o que caracteriza o diagnóstico. Tal fato denota a importância do oftalmologista tanto na investigação inicial quanto no seguimento de afecções sistêmicas.
Palavras-chave: Distrofias Retinianas; Retinose Pigmentar; Síndrome de Bardet-Biedl.
RESUMEN
INTRODUCCIÓN: El síndrome de Bardet-Biedl es una enfermedad de herencia autosómica recesiva rara cuyas principales manifestaciones clínicas son polidactilia, obesidad, retardo mental, hipogonadismo y distrofia retiniana que causa progresiva pérdida visual desde la infancia.
REPORTE DE CASO: Paciente, 17 años, sexo masculino, presentó baja acuidad visual progresiva en ambos ojos. Presentaba déficit intelectual y malformación cardíaca desde el nacimiento, así como estrabismo divergente. Además, tenía baja estatura, obesidad y polidactilia en manos y pies. Al examen oftalmológico, se constató acuidad visual con corrección de 0,1 por la tabla de Snellen en ambos ojos, presión intraocular y biomicroscopía normales. Al realizarse la fundoscopía, se ha observado decoloración del epitelio pigmentado de la retina, lo que le daba al fondo una coloración agrisada y un aspecto moteado, disminución del calibre vascular, presencia de palidez papilar, emigración de pigmento en calidad de espículas óseas, incluso macular, y aumento del reflejo foveal.
CONCLUSIÓN: la distrofia retiniana está presente en cerca del 90% de los casos ya descritos en el síndrome de Bardet-Biedl y muchas veces es lo que caracteriza el diagnóstico. Tal hecho denota la importancia del oftalmólogo tanto en la averiguación inicial como en el seguimiento de afecciones sistémicas.
Palabras-clave: Distrofias Retinianas; Retinitis Pigmentosa; Síndrome de Bardet-Biedl.
INTRODUCTION
Bardet-Biedl syndrome (BBS) is a rare autosomal recessive disease with a prevalence of 1:160,000-1:13,500.1 The chief clinical manifestations are retinal dystrophy, polydactylism, abdominal obesity, mental retardation, and hypogenitalism. Retinal dystrophy is the most common finding and causes progressive visual loss since childhood2. Here we describe the case of a patient with clinical manifestations compatible with the diagnosis of BBS.
CASE REPORT
A 17-year-old male Caucasian student from Belo Horizonte, Brazil, was referred to the Institute of Ophthalmology Medical Sciences (Instituto de Olhos Ciências Médicas) owing to a history of progressive low visual acuity in both eyes. The patient was reported to have intellectual disability and cardiac malformation since birth as well as strabismus, for which he used an eye patch during childhood (Figure 1).
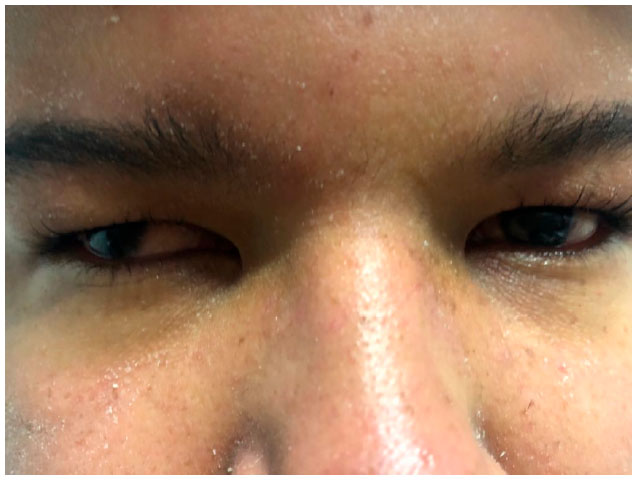
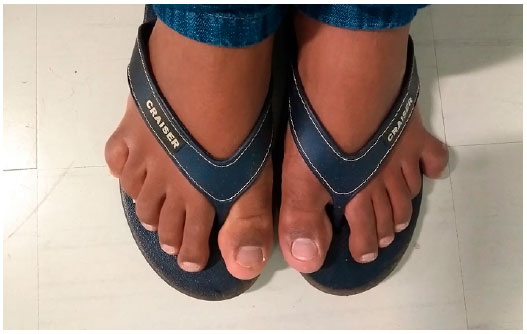
Renal function and levels of serum sodium and potassium, glucose, testosterone, and follicle-stimulating and luteinizing hormones were normal. His parents were non-consanguineous, and family history revealed that a maternal cousin had died of an unreported cardiac issue. Physical examination revealed short stature, obesity, and polydactyly of hands and feet (Figures 2 and 3).

Ophthalmologic examination using Snellen chart revealed visual acuity of 0.1 in both eyes. He had normal intraocular pressure and biomicroscopy result. Fundoscopy revealed paleness of the optic papilla, changes in the retinal pigment epithelium (RPE), and reduction of vessel diameter in both eyes. The reliability of the visual field test was low in both eyes.
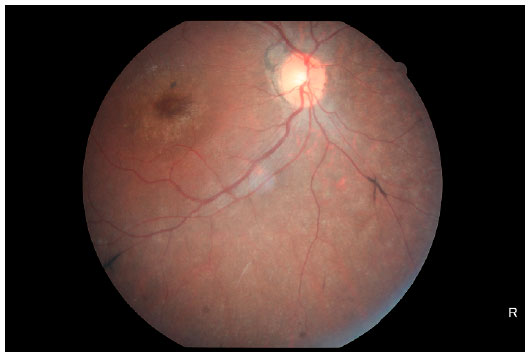
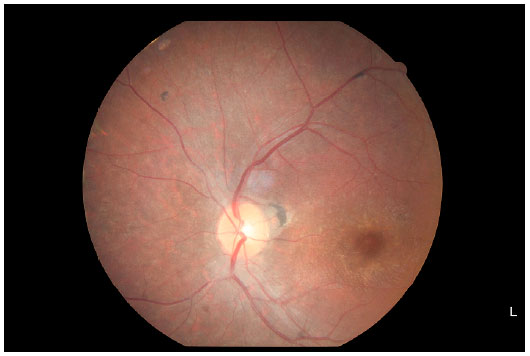
Retinography (Figures 4 and 5) showed relevant characteristics of tapetoretinal degeneration, such as discoloration of RPE, giving the fundus a grayish color and mottled appearance; reduced vessel diameter; macular and peripheral bone spicule-shaped pigment deposits; and increased foveal reflex.
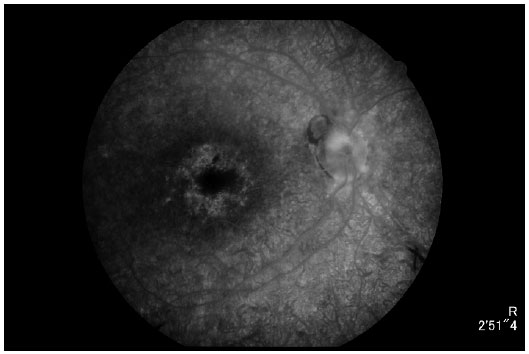
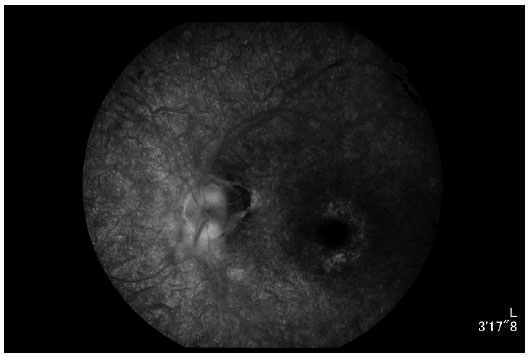
Angiography (Figures 6 and 7) revealed diffuse hyperfluorescence, suggesting a decreased number of RPE cells; hypofluorescence by peridiscal and perivascular block, suggesting pigment mobilization; and hyperfluorescence due to a window defect, suggesting perifoveal RPE atrophy.
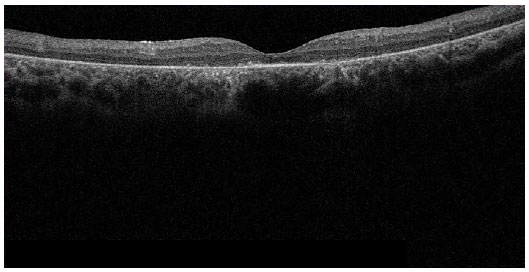
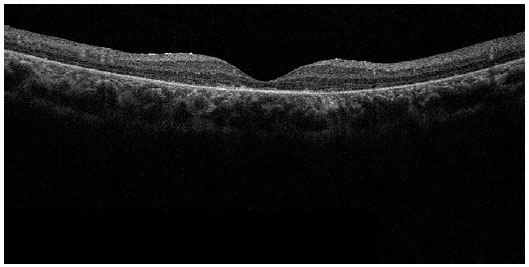
Optical coherence tomography (Figures 8 and 9) showed diffuse thinning of the retina, mainly in the outer retinal layer, and diffuse loss of photoreceptors.
DISCUSSION
BBS is an autosomal recessive genetic disorder. It is considered to be a ciliopathy wherein the genes involved in primary ciliary function are affected3. It has high genetic heterogeneity, and at least 20 genes have been mapped on different chromosomes2.
Primary characteristics include cone-rod dystrophy and loss of vision since childhood, along with nocturnal blindness; post-axial polydactylism; abdominal obesity; intellectual disability; male hypogenitalism and other complex malformations of the female genitourinary tract; and renal dysfunction, which is a major cause of morbidity and mortality3.
Secondary characteristics include ocular anomalies, such as strabismus, cataract, and astigmatism; speech disorder/delay; brachydactyly or syndactyly; developmental delay; ataxia; diabetes mellitus; craniofacial dysmorphism; nephrogenic diabetes insipidus; hepatic fibrosis; and congenital heart disease. Beales et al.7 reported that the diagnosis is confirmed by the presence of either four primary characteristics or three primary and two secondary characteristics. Complications such as hypothyroidism, Hirschsprung’s disease, epilepsy, genital anomalies, anal stenosis, and abnormal dentition are rare2.
Molecular genetic testing using clinically available kits can be used to confirm the diagnosis. Genotype-phenotype correlations are not evident; therefore, no single gene test is recommended. Therefore, it is prudent to sequence genes with high rates of mutation2.
The diagnosis of BBS can be delayed owing to the slow onset and variable clinical manifestations1. In addition, there are other syndromes that present similar combinations of complications such as ocular defects, mental retardation, genital hypoplasia, obesity, and digital anomalies. Although less common, Laurence-Moon syndrome, Alstrom syndrome, and Biemond II syndrome should be considered in the differential diagnosis2.
The most common clinical manifestation, which is used to confirm the diagnosis, is the development of retinal dystrophy2,4, wherein the function of rods and cones is affected, culminating in the atypical presentation of retinitis pigmentosa with early macular involvement. Maculopathy may or may not be associated with peripheral retinal degeneration5,6.
Early signs of retinal dysfunction may not be apparent until the age of 7-8 years, when night blindness insidiously occurs7. By the second to third decade of life, the macula is impaired in all affected individuals, accompanied by visual acuity of 0.1 or worse. Reportedly, 63.6% of the affected individuals are legally blind at the age of 20 years8.
Visual fields are usually abnormal at the age of 10 years and progress with a visual loss of up to 3°/year. A little more than one central island of vision usually remains at the age of 20 years1.
In most cases, electroretinography (ERG) result is abnormal and typically shows residual or absent responses, with high thresholds of adaptation to the dark. Progressive retinal degeneration and low reliability of the visual field test in this syndrome are responsible for the lack of correlation between the maximum ERG response and the small area of the visual field. Although ERG result may be abnormal up to the age of 14 months, significant cone-rod dystrophy is not apparent in most patients aged <5 years, and the agreement with the results of ERG test at this age is often poor. Therefore, unless strongly indicated, ERG test should be postponed until the age of at least 4 years2.
Other ophthalmologic findings include nystagmus, strabismus, high myopia, cataract, and glaucoma2.
Post-axial polydactylism occurs in more than 90% of cases and may be the only dysmorphic sign at birth. This complication may affect the limbs, the ulnar sides of the arms, or the fibular sides of the legs and may be accompanied by brachydactyly and/or partial syndactyly (usually between the second and third toes)5.
Abdominal obesity is moderate in approximately 90% of patients, which was also observed in our patents1. This complication appears in childhood and persists in adulthood; it should be managed with diet, exercise, and behavioral therapies.
Hypogenitalism is common in men possibly because of a primary genetic defect4.
Abnormalities of glucose metabolism, such as carbohydrate intolerance and type I and type II diabetes mellitus, have been reported in patients with BBS. Therefore, patients with BBS should be screened for changes in glucose metabolism.
Renal dysfunction is the leading cause of morbidity and mortality in BBS. Therefore, monitoring the patient’s renal function is necessary2.
In summary, BBS can be detected on the basis of clinical manifestations without the need for laboratory tests. Nephrologists can help in the diagnosis of BBS in patients with renal dysfunction. BBS cases in dialysis units may be used to screen the syndrome and consequently provide more data on the prevalence of BBS in developing countries, such as Brazil, where data on the incidence of this syndrome are limited.
Because BBS is an autosomal recessive disease, genetic counseling is fundamental for patients and their families2. A few studies have reported cases of female patients who have given birth despite the high rate of infertility in BBS; therefore, family planning is vital2,4.
CONCLUSION
The primary clinical manifestations of BBS include retinal dystrophy, post-axial polydactylism, abdominal obesity, intellectual disability, male hypogenitalism, complex malformations of the female genitourinary tract, and renal dysfunction.
We described the case of a patient with three primary characteristics (i.e., polydactylism, intellectual disability, and retinal dystrophy) and at least two secondary characteristics (i.e., cardiac malformation, developmental delay, and strabismus) for the diagnostic confirmation. Retinal dystrophy occurs in approximately 90% of the cases and usually confirms the diagnosis. This manifestation indicates the importance of the role of an ophthalmologist in the initial investigation and follow-up of systemic complications.
REFERENCES
1. Lavinsky J, Goldhardt R, Ariente SK, Domingues CG, Lavinsky F. Síndrome de Bardet-Biedl: relato de dois casos. Arq Bras Oftalmol. 2003; 66(5). Acessado em: 28/10/2018. Disponível em: http://www.scielo.br/pdf/%0D/abo/v66n5/18160.pdf.
2. Forsythe E, Beales PL. Bardet-Biedl syndrome. 2003 Jul (Updated 2015 Apr 23). In: Adam MP, Ardinger HH, Pagon RA, et al. (ed.). GeneReviews® (Internet). Seattle: University of Washington; 1993-2018. Acessado em: 28/10/2018. Disponível em: https://www.ncbi.nlm.nih.gov/books/NBK1363/.
3. Mockela A, Perdomo Y, Stutzmann F, Letsch J, Marion V, Dollfus H. Retinal dystrophy in Bardet-Biedl syndrome and related syndromic ciliopathies. Progress in Retinal and Eye Research. 2011; 30(4). Acessado em: 28/10/2018. Disponível em: https://www.sciencedirect.com/science/article/pii/S1350946211000164.
4. Forsythe E, Beales PL. Bardet-Biedl syndrome. Eur J Human Gen. 2013; 21. Acessado em: 28/10/2018. Disponível em: https://www.nature.com/articles/ejhg2012115.
Funding: No specific financial support was available for this study
CEP Approval: Not applicable
Disclosure of potential conflicts of interest: None of the authors have any potential conflict of interest to disclose
Received on:
October 8, 2018.
Accepted on:
November 7, 2018.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados



 Ler em português
Ler em português
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket