

Arthur Borges dos Santos1; Antônio Bandeira e Silva2; Mateus Abdalla Bastos3; Daniel de Almeida Wanderley Guedes4; Kátia Delalibera Pacheco5; Fabrício Tadeu Borges6; Marcos Pereira de Ávila7
DOI: 10.17545/eoftalmo/2018.0035
ABSTRACT
Retinal pigment epithelium pattern dystrophies comprise a heterogeneous group of hereditary diseases of the macula that originate from the mutations of the peripherin/RDS gene. They are characterized by the accumulation of lipofuscin in the retinal pigment epithelium, which results in different phenotypical patterns and may be confused with drusen. In certain cases, the course is not favorable, due to geographic atrophy formation or choroidal neovascularization. Here, we report a case of pattern dystrophy of the retinal pigment epithelium in a male patient with poor vision, associated with retinal pigment epithelium detachment and external retinal tubulation in the fovea of the left eye. The main differential diagnosis was age-related macular degeneration.
Keywords: Retinal Dystrophies; Retinal Pigment Epithelium; Macular Degeneration.
RESUMO
As distrofias em padrão do epitélio pigmentar da retina constituem um grupo heterogêneo de doenças hereditárias da mácula que têm origem em mutações do gene periferina/RDS. São caracterizadas pelo acúmulo de lipofucsina no epitélio pigmentar da retina que resultam em diferentes padrões fenotípicos e podem ser confundidas com drusas. Em alguns casos o curso não é favorável pois há formação de atrofia geográfica ou neovascularização de coroide. Apresentamos um caso de distrofia padrão do epitélio pigmentar da retina em um paciente do sexo masculino com baixa visão associada a descolamento de epitélio pigmentar da retina e tubulação da retina externa em fóvea do olho esquerdo cujo principal diagnóstico diferencial foi realizado com a degeneração macular relacionada à idade.
Palavras-chave: Distrofias Retinianas; Epitélio Pigmentado da Retina; Degeneração Macular.
RESUMEN
Las distrofias en calidad del epitelio pigmentario de la retina constituyen un grupo heterogéneo de enfermedades hereditarias de la mácula que tienen origen en mutaciones del gene periferina/RDS. Se caracterizan por el acúmulo de lipofucsina en el epitelio pigmentario de la retina que resultan en diferentes estándares fenotípicos y se pueden confundir con drusas. En algunos casos, el curso no es favorable, pues hay formación de atrofia geográfica o neovascularización coroidea. Presentamos un caso de distrofia estándar del epitelio pigmentario de la retina en un paciente del sexo masculino con baja visión asociada al desprendimiento de epitelio pigmentario de la retina y tubulación de la retina externa en fóvea del ojo izquierdo, cuyo principal diagnóstico diferencial ha sido realizado con la degeneración macular relacionada a la edad.
Palabras-clave: Distrofias Retinianas; Epitelio Pigmentado de la Retina; Degeneración Macular.
INTRODUCTION
Retinal pigment epithelium pattern dystrophies (PD) are a group of diseases with dominant autosomal inheritance, associated with mutations in the peripherin/RDS gene and characterized by an accumulation of lipofuscin in the retinal pigment epithelium (RPE) or in the subretinal space1,2.
The arrangement of this pigment buildup defines the clinical phenotypes, but often more than one presentation may coexist3. The clinical presentation has similarities with other diseases of the macula, such as age-related macular degeneration (ARMD), Stargardt disease, and central serous chorioretinopathy (CSCR)2,4.
Patients with PD may remain asymptomatic throughout life or present visual symptoms that range from mild visual impairment and metamorphopsia to loss of central vision due to the formation of geographic atrophy or to choroidal neovascularization.
We present herein a case of pattern dystrophy in which the main differential diagnosis was ARMD.
CASE REPORT
A male patient, white, aged 46 years, complained of gradual visual blurring in the left eye with onset 6 months prior, but with partial spontaneous improvement observed in the last 2 months. On ophthalmological examination, he presented a corrected visual acuity of 20/20 in the right eye and 20/60 in the left eye. There were no changes in the biomicroscopic examination and the intraocular pressure was 14 mmHg in both eyes. Fundoscopy of both eyes showed yellowish subretinal deposits (flecks) distributed in the posterior pole and the middle periphery, with an apparent confluence in the central region of the macula (Figures 1A, 1B). In the fovea of the left eye, a rounded, raised lesion was seen, with hypopigmentation of the RPE (Figure 1B). The autofluorescence examination showed multiple hyperautofluorescent areas, corresponding to the sites of the deposits (Figures 1C, 1D). Fluorescein angiography presented diffuse areas of late hyperfluorescence due to impregnation, with greater intensity in the macular region of both eyes (Figures 1E, 1F). Spectral-domain optical coherence tomography (SD-OCT) of the right eye showed the presence of hyperreflective material between the neurosensory retina and the RPE-Bruch’s membrane complex (Figure 1G), while in the left eye, dislocation of the RPE and outer retinal tubulation in the foveal region were observed (Figure 1H), The diagnostic investigation was complemented with an angiography examination by OCT, due to the possibility of the existence of choroidal neovascularization at the PD site in the left eye, but no signs of flow were evidenced in the angiogram (Figure 2). A full-field electroretinogram showed retinal dysfunction of cones and rods in both eyes, and the electro-oculogram was sub-normal. No genetic test was conducted to check for a mutation of the peripherin/RDS gene. The patient was advised to have regular clinical follow-up and weekly tests with the Amsler grid.
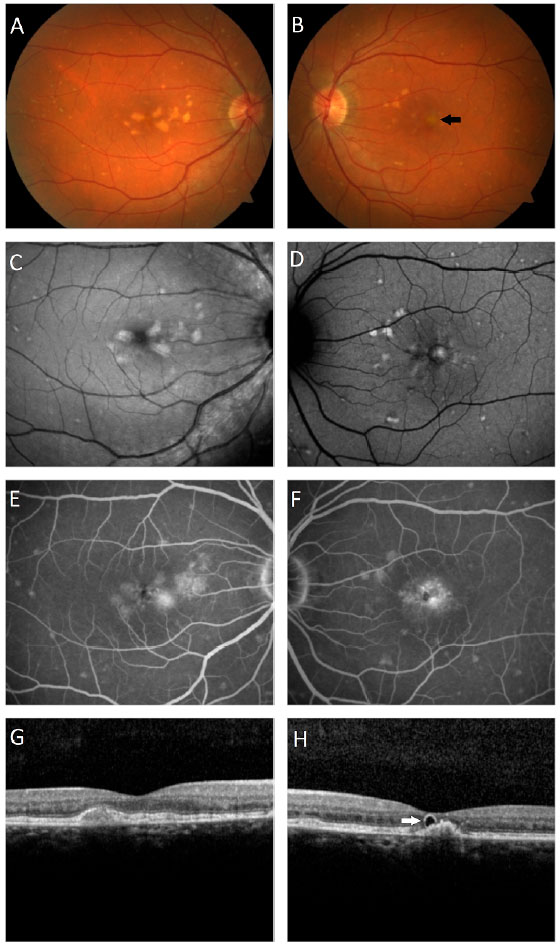
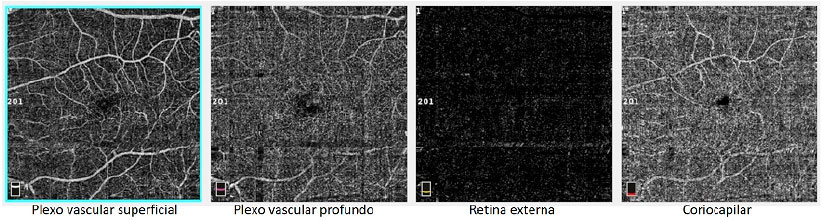
DISCUSSION
Pattern dystrophies are a heterogenous group of macular diseases, generally diagnosed around the 2nd or 3rd decades of life. They may present similarities with other macular conditions. Among these, ARMD is one of the most frequent causes of diagnostic confusion and unnecessary treatments in these patients1,2,5. This is due to the appearance of the subretinal deposits, which are often confused with drusen, and also to the possibility of areas of RPE hypopigmentation/atrophy or choroidal neovascularization appearing in older patients6. In a study conducted by Ozkaya et al., only 45% of patients were given a correct diagnosis of PD; the remaining 55% were diagnosed as having ARMD, CSCR, or nonspecific RPE changes2.
The clinical findings that differentiate pattern dystrophies from ARMD include an early age of onset of the disease, the absence of drusen in their characteristic format, and the presence of greyish-yellow pigments in various patterns in the macular area7. This material, composed of lipofuscin, originates from a phagocytic dysfunction of the RPE next to the external segments of the photoreceptors, determined by a mutation in the peripherin/RDS gene, located on chromosome6,4,8.
In PD, the autofluorescence examination shows areas of hyperautofluorescence in the sites of pigment accumulation, and hypoautofluorescence in atrophic areas of the RPE9. Fluorescein angiography is useful for investigation of choroidal neovascularization, and for differentiating pattern dystrophy from Stargardt disease, as there is no choroidal silence sign3,10. On OCT examination, a disorganization of cell architecture may be identified in the external layers of the retina, as well as a thickening of the RPE and the presence of hyperreflective material, corresponding to lipofuscin deposits, located in the RPE or between the neurosensory retina and the RPE-Bruch’s membrane complex2,11. The results of the electro-oculogram and electro-retinogram, particularly multifocal in the latter case, may exhibit changes6,12,13.
Although the majority of cases of pattern dystrophy present good visual prognosis, loss of central vision at advanced ages may occur in up to 50% of cases, caused by the occurrence of geographic atrophy or choroidal neovascularization8,13,14. If the formation of a neovascular membrane occurs, the treatment involves the use of anti-VEGF agents, either in isolation or in combination with photodynamic therapy (PDT), but there are case reports of spontaneous remission of the neovascularization without the need for treatment6,7,8,13.
In the presently described case, the patient exhibited characteristic findings of multifocal PD, with an aspect similar to fundus flavimaculatus in both eyes. This is characterized by the. Presence of fleck-like yellowish deposits in the posterior pole and beyond the vascular arcades4,15. In the right eye, it was also possible to visualize the butterfly-shaped subtype, due to the presence of deposits arranged in wing-shaped projections, spreading out from the foveal center1,16. On fundoscopy, an initial diagnosis of ARMD was suggested by the visual aspect of the deposits in the macular region, similar to soft confluent drusen in both eyes, as well as by the foveal hypopigmentation with a raised RPE in the left eye, and by the presence of outer retinal tubulation in the OCT of the left eye. This initial diagnostic hypothesis was ruled out because the patient presented a younger age than is compatible with the disease, the lesions in the middle periphery differed in clinical appearance from drusen, and in the SD-OCT, the deposits were located predominantly in the subretinal space. The patient’s report of poor vision in the left eye, with partial improvement of the symptoms in the previous few months, together with the analysis of the OCT findings in this eye’s foveal region, suggested the presence of choroidal neovascularization at the site, but this hypothesis was ruled out after the angiography by OCT. The formation of the PD, therefore, may be related to the presence of sub-RPE vitelliform material17, while the presence of outer retinal tubulation is a consequence of previous lipofuscin damage to the photoreceptors, with subsequent reabsorption of the deposit18.
This report adds to the literature a case of PD in the RPE that featured more than one subtype of the disease and whose main differential diagnosis was ARMD. The SD-OCT also revealed the presence of outer retinal tubulation in the foveal region of the left eye, a finding that is rarely described in the literature in cases of PD, but more frequently for ARMD. It is concluded that when PD in the RPE is suspected, it is necessary to use the diagnostic tools available to avoid diagnostic confusion with other diseases of greater prevalence and unnecessary treatments.
REFERENCES
1. Isaac DLC, Santos RAV, Ávila M. Distrofia em forma-de-borboleta: relato de caso. Arq Bras Oftalmol. 2007; 70(1):129-32.
2. Ozkaya A, et al. Clinical and imaging findings of pattern dystrophy subtypes; Diagnostic errors and unnecessary treatment in clinical practice. J Fr Ophtalmol. 2018; 41(1):21-9.
3. Houly JR. Pattern dystrophy of retinal pigment epithelium. Eye Care Vis. 2017; 1(1):1-2.
4. Silva R, Farah ME. Manual de retina. Lisboa: Lidel Edições Técnicas; 2015.
5. Marmor MF, McNamara A. Pattern dystrophy of the retinal pigment epithelium and geographic atrophy of the macula. Am J Ophthalmol. 1996; 122(3):382- 92.
6. Parodi MB, Pozzo S, Ravalico G. Photodynamic therapy for choroidal neovascularization associated with pattern dystrophy. Retina. 2003; 23(2):171-6.
7. Anastasakis A, et al. Spontaneous regression of choroidal neovascularization in a patient with pattern dystrophy. Case Rep in Ophthalmol Med. [online journal]. 2016 [Acessado em: 02/06/2018]; 2016: 9685290 [about 5p.]. Disponível em: https://www.hindawi.com/journals/criopm/2016/9685290/.
8. Empeslidis T, Vardarinos A, Deane J, Banerjee S. Intravitreal ranibizumab in the treatment of butterfly-shaped pattern dystrophy associated with choroidal nevascularization: a case report. Case Rep Ophthalmol. 2012; 3(1):77-82.
9. Boon CJF, Klevering BJ, Keunen JEE, Hoyng CB, Theelen T. Fundus autofluorescence imaging of retinal dystrophies. Vision Res. 2008; 48(26):2569-77.
10. Boon CJF, Schooneveld MJ, Hollander A, Lith-Verhoeven JJC, Zonneveld-Vrieling MN, Theelen T, et al. Mutations in the peripherin/RDS gene are an important cause of multifocal pattern dystrophy simulating STGD1/fundus flavimaculatus. Br J Ophthalmol. 2007; 91(11): 1504-11.
11. Hannan SR, Salvo G, Stinghe A, Shawkat F, Lotery AJ. Common spectral domain OCT and electrophysiological findings in diferent pattern dystrophies. Br J Ophthalmol. 2013; 97(5):605-10.
12. Duinkerke-Eerola KU, Pinckers A, Cruysberg JRM. Pattern dystrophy of the retinal pigment epithelium. Int Ophthalmol. 1987; 11(2):65-72.
13. Parodi MB, Iacono P, Cascavilla M, Zucchiatti I, Kontadakis DS, Bandello F. Intravitreal bevacizumab for subfoveal choroidal neovascularization associated with pattern dystrophy. Invest Ophthalmol Vis Sci. 2010; 51(9):4358-61.
14. Francis PJ, Schultz DW, Gregory AM, Schain MB, Barra R, Majewski J, et al. Genetic and phenotypic heterogeneity in pattern dystrophy. Br J Ophthalmol. 2005; 89(9):1115-9.
15. Roy R, Kumar S, Chandrasekharan, Ghose A, Sharma P. Multimodal imaging in multifocal pattern dystrophy simulating fundus flavimaculatus. Indian J Ophthalmol. 2016; 64(5):395-6.
16. Saatci AO, Yasti ZO, Köse S, Memişoglu B. Butterfly-like pattern dystrophy and unilateral choroidal neovascularization. Acta Ophthalmol Scand. 1998; 76(6):734-6.
17. Grob S, Yonekawa Y, Eliott D. Multimodal imaging of adult-onset foveomacular vitelliform dystrophy. Saudi J Ophthalmol. 2014; 28(2):104-10. 18. Goldberg NR, Greenberg JP, Laud K, Tsang S, Freund KB. Outer Retinal Tubulation in degenerative retinal disorders. Retina. 2013; 33(9):1871-6.







Funding: No specific financial support was available for this study
CEP Approval: Not applicable
Disclosure of potential conflicts of interest: None of the authors have any potential conflict of interest to disclose
Received on:
August 21, 2018.
Accepted on:
November 6, 2018.
 eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
eOftalmo está licenciada com uma Licença Creative Commons Atribuição-NãoComercial 4.0 Internacional.
![]() © 2026 Todos os Direitos Reservados
© 2026 Todos os Direitos Reservados
 Ler em português
Ler em português
 Português PDF
Português PDF
 Imprimir
Imprimir
 Enviar este artigo por email
Enviar este artigo por email
 Como citar este artigo
Como citar este artigo
 Enviar um comentário
Enviar um comentário
 Mendeley
Mendeley
 Pocket
Pocket